Dynamical Responses in Energetic Materials
Micro- and mesoscale (below 1 μm) phenomena play a dominant role in the resulting macroscopic properties for a wide variety of material types, e.g., soft matter, biological matter, complex fluids, composite materials or additively-manufactured materials. Predictive modelling and simulation of materials response requires multiscale modelling at length and time scales ranging from atomistic to continuum. Modelling and simulation at this scale is far from amenable to atomistic scale approaches, while continuum scale simulations lack the fidelity to properly include material microstructure and rely on phenomenological models to effectively recover and/or estimate the physical processes occurring at the micro and mesoscales. Thus, a key gap in the theoretical foundation and computational methods exists at the microscale/mesoscale across various scientific fields, where studies have been performed over an extensive scope of materials and applications, including, but not limited to, the life sciences (proteins, colloidal suspensions, bio-membranes, and micelles), industrial applications (surfactants, asphaltenes, and viscoelastic fluids), national defence applications (energetic material composites and liquid propellants), and novel materials (self-assembled block copolymers/nanoparticles). We are developing novel particle-based mesoscale modelling tools for predicting the spatial-temporal energy release, heat transfer, mass transfer and chemical reactivity of energetic materials. We aim to establish a rigorous, well-founded, and general computational approach for systematic study directed toward identifying and characterising fundamental aspects of the dynamic response of energetic materials.
Nanoconfined Fluids
The physics of fluids in nanoconfinement such as micropores or at interfaces with solid surfaces such as graphene differs significantly from the physics in the bulk. The significant and often counterintuitive changes in fluid behaviour in nanospace are caused by dominant effects of interactions between the fluid molecules and surface atoms. Results of strong fluid-solid interactions are inhomogeneities in fluid density, appearance of specific fluid phases, slow-down of fluid diffusion and transport, e.g. aqueous solutions in hydrophilic mineral micropores, or speed-up of fluid diffusion and transport, e.g. water in carbon nanotubes. We use molecular dynamics and Monte Carlo simulations to understand, describe and quantify at the molecular level effects of nanospace and fluid-solid interactions on the phase, structural, diffusion and transport behaviour of nanoconfined fluids. A molecular-level understanding of nanoconfined fluid behaviour plays a critical role in rational design of porous materials or in development of graphene-based technologies.
Interfacial Phenomena at Nanoscopically Heterogenous Surfaces
There is a lot of unambiguous experimental evidence that modification of solid surfaces may dramatically alter the structure and phase behaviour of the ambient fluid. It is already well known that geometrical or chemical modification of solid surfaces can induce completely new types of phase transitions and critical phenomena, related to but distinct from wetting occurring at planar (and ideally smooth) surfaces. Using methods of statistical mechanics (Density Functional Theory, Effective Hamiltonian Models, Scaling,…) we endeavour to capture the essential physics of these phenomena and understand the subtle interplay between the surface geometry, molecular interactions, interfacial fluctuations and measureable characteristics of the system (such as adsorption). Our main focus is on surfaces whose heterogeneity is characterized by a nanoscopically small length-scale, a research area which is still in its infancy and revealing a number of counter-intuitive phenomena that cannot be captured by standard tools. Beside its theoretical importance, the research has also tight connections with modern nanotechnologies dealing with fabrication of functional materials or for a development of devices controlling flows of extremely tiny volumes of liquid, such as “lab-on-a-chip”.
Using dissipative particle dynamics to model polymeric systems
 This chapter reviews the principles and applications of dissipative particle dynamics (DPD) as a coarse‑grained simulation technique for polymeric systems. It begins by outlining the multiscale structural and dynamical complexity of polymers, which spans many orders of magnitude in time and length and makes fully atomistic simulations impractical. DPD is introduced as a mesoscale, momentum‑conserving method in which soft beads represent groups of atoms or Kuhn segments and interact via conservative, dissipative, and random forces chosen to preserve hydrodynamics and impose a thermostat. The Groot-Warren formulation is described in detail, including soft repulsive bead-bead interactions, spring and FENE bond potentials for flexible and semiflexible chains, and additional segment-segment repulsions to suppress unphysical chain crossing in entangled melts. Electrostatic interactions in ion‑containing polymers are then incorporated through smeared charge distributions and Ewald‑type treatments, with discussion of charge assignment strategies for quenched and annealed polyelectrolytes and the delicate balance between Coulombic and excluded‑volume forces. Parametrization strategies are presented that map DPD repulsion parameters onto Flory-Huggins interaction parameters and experimental compressibilities, with extensions to mixtures of beads of different sizes and densities. The second part of the chapter surveys DPD applications: polymer solutions and melts across concentration regimes; self‑assembly of block copolymers into micelles, vesicles, and complex nanoparticles; electrostatic co‑assembly of oppositely charged polyelectrolytes into interpolyelectrolyte complexes; and polymer behavior at interfaces and under 1D-3D confinement, including adsorption, slit pores, and emulsion droplets. The authors close by highlighting both the power and limitations of DPD, emphasizing the need for physically sound coarse‑graining and parametrization, and pointing to future developments such as improved electrostatics, adaptive‑resolution schemes, and data‑driven parameter optimization to extend DPD’s role in designing advanced polymeric materials.
This chapter reviews the principles and applications of dissipative particle dynamics (DPD) as a coarse‑grained simulation technique for polymeric systems. It begins by outlining the multiscale structural and dynamical complexity of polymers, which spans many orders of magnitude in time and length and makes fully atomistic simulations impractical. DPD is introduced as a mesoscale, momentum‑conserving method in which soft beads represent groups of atoms or Kuhn segments and interact via conservative, dissipative, and random forces chosen to preserve hydrodynamics and impose a thermostat. The Groot-Warren formulation is described in detail, including soft repulsive bead-bead interactions, spring and FENE bond potentials for flexible and semiflexible chains, and additional segment-segment repulsions to suppress unphysical chain crossing in entangled melts. Electrostatic interactions in ion‑containing polymers are then incorporated through smeared charge distributions and Ewald‑type treatments, with discussion of charge assignment strategies for quenched and annealed polyelectrolytes and the delicate balance between Coulombic and excluded‑volume forces. Parametrization strategies are presented that map DPD repulsion parameters onto Flory-Huggins interaction parameters and experimental compressibilities, with extensions to mixtures of beads of different sizes and densities. The second part of the chapter surveys DPD applications: polymer solutions and melts across concentration regimes; self‑assembly of block copolymers into micelles, vesicles, and complex nanoparticles; electrostatic co‑assembly of oppositely charged polyelectrolytes into interpolyelectrolyte complexes; and polymer behavior at interfaces and under 1D-3D confinement, including adsorption, slit pores, and emulsion droplets. The authors close by highlighting both the power and limitations of DPD, emphasizing the need for physically sound coarse‑graining and parametrization, and pointing to future developments such as improved electrostatics, adaptive‑resolution schemes, and data‑driven parameter optimization to extend DPD’s role in designing advanced polymeric materials.
- K. Procházka, K. Šindelka, Z. Limpouchová, M. Lísal: Using dissipative particle dynamics to model polymeric systems. Computational Methods for the Multiscale Modeling of Soft Matter, Part One - Soft matter modeling methods, pp. 3-35, edited by P. Carbone and N. Clarke; Methods in Molecular and Materials Modeling Book Series, series editor R. Catlow; Elsevier, 2026. ISBN: 978-0-443-27314-8. DOI
Interpretable machine-learning enhanced parametrization methodology for Pluronics–water mixtures in DPD simulations
 Dissipative particle dynamics (DPD) is an incredibly powerful tool for simulating the behavior of structured fluids. However, identifying the appropriate model parameters to accurately replicate physical properties remains a challenge. This study showcases the benefits of integrating machine learning techniques into the top-down parameterization of Pluronic systems. The proposed workflow outlines a data-driven approach to accurately determine model parameters tailored to various Pluronic systems. Gaussian process regression (GPR)-based surrogate models effectively replicate the results of DPD simulations, delivering faster responses that streamline parameter optimization and enable the calibration of Pluronic systems against experimental data. Although DPD simulations provide valuable insight, their high computational cost, due to extensive simulations and post-processing, presents a challenge. The GPR-based surrogate model addresses this by modeling the relationships between input parameters and output properties. SHAP (SHapley additive exPlanations) analysis enhances model interpretability, providing deeper insights into the relationships and causal mechanisms between the input parameters and the predicted properties. The combination of GPR and SHAP analysis provides an interpretable machine learning approach, enabling a more efficient optimization process and reducing the need for exhaustive simulations. This work lays a foundation for generalizing the parameterization process across Pluronic systems and conditions, such as varying temperatures, by incorporating additional DPD model input parameters.
Dissipative particle dynamics (DPD) is an incredibly powerful tool for simulating the behavior of structured fluids. However, identifying the appropriate model parameters to accurately replicate physical properties remains a challenge. This study showcases the benefits of integrating machine learning techniques into the top-down parameterization of Pluronic systems. The proposed workflow outlines a data-driven approach to accurately determine model parameters tailored to various Pluronic systems. Gaussian process regression (GPR)-based surrogate models effectively replicate the results of DPD simulations, delivering faster responses that streamline parameter optimization and enable the calibration of Pluronic systems against experimental data. Although DPD simulations provide valuable insight, their high computational cost, due to extensive simulations and post-processing, presents a challenge. The GPR-based surrogate model addresses this by modeling the relationships between input parameters and output properties. SHAP (SHapley additive exPlanations) analysis enhances model interpretability, providing deeper insights into the relationships and causal mechanisms between the input parameters and the predicted properties. The combination of GPR and SHAP analysis provides an interpretable machine learning approach, enabling a more efficient optimization process and reducing the need for exhaustive simulations. This work lays a foundation for generalizing the parameterization process across Pluronic systems and conditions, such as varying temperatures, by incorporating additional DPD model input parameters.
- N. Lauriello, D.N. Ponnana, Z. Ma, K. Šindelka, A. Buffo, G. Boccardo, D. Marchisio, W. Pan: Interpretable machine-learning enhanced parametrization methodology for Pluronics–water mixtures in DPD simulations, Soft Matter 21, 5833-5851, 2025. DOI
Dissipative particle dynamics as a computational tool to detect the morphology-rheology interplay in Pluronic F68/water mixtures: A promising drug carrier
Pluronics, also known as poloxamers, are amphiphilic triblock copolymers widely employed in drug delivery systems due to their tunable self-assembly and biocompatibility. Among them, Pluronic F68 (Poloxamer 188) exhibits thermoresponsive behavior in aqueous solution, forming ordered supramolecular structures at high concentrations and temperatures. In this work, we investigate the morphological and rheological properties of a 45 wt% Pluronic F68 aqueous system at different temperatures through a combination of experimental and computational approaches. Rheological measurements and Small-Angle X-ray Scattering (SAXS) confirm the formation of a body-centered cubic (BCC) structure at higher temperatures and highlight the emergence of viscoelastic solid-like behavior. To support and extend these findings, Dissipative Particle Dynamics (DPD) simulations are employed to model the nanostructure evolution and the impact of temperature on self-assembly and material properties. This integrated approach provides a consistent framework to characterize the temperature-induced transition from fluid-like to solid-like states and sets the groundwork for future simulation studies incorporating drug cargo. The results offer valuable insights into the design of thermoresponsive drug delivery systems and demonstrate the potential of DPD in capturing complex structure–property relationships in amphiphilic polymer systems.

- N. Lauriello, N. A. Di Spirito, K. Šindelka, G. Boccardo, R. Pasquino, N. Grizzuti, D. Marchisio: Dissipative particle dynamics as a computational tool to detect the morphology-rheology interplay in Pluronic F68/water mixtures: A promising drug carrier. J. Colloid Interface Sci. 700, 138525, 2025. DOI
Confined active particles: Wall accumulation and correspondence between active and fluid systems
Active Brownian particles (ABPs) are a generic model for synthetic active matter systems, such as active colloids, characterized by their ability to self-propel while also exhibiting Brownian motion. Under confinement, ABPs display propulsion-induced wall accumulation, a behavior that can be harnessed for microfluidic and lab-on-a-chip applications. We investigate ABPs confined in slit pores using overdamped Langevin dynamics and study their wall accumulation as a function of particle activity and slit width, at both gas and liquid densities. Furthermore, we examine the correspondence between confined ABPs and their equilibrium counterparts, confined fluid particles capable of completely wetting the slit walls.
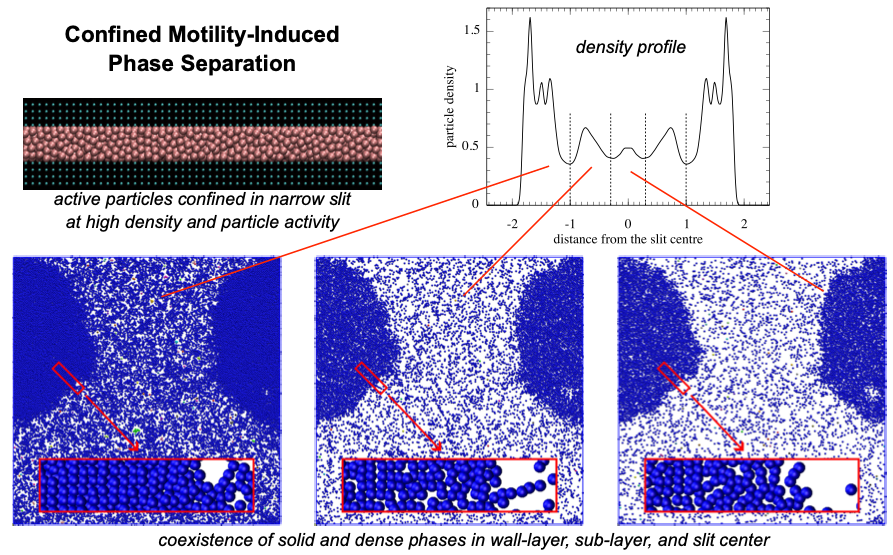
- K. Šindelka, A. Gadermeteva, M. Lísal: Confined active particles: Wall accumulation and correspondence between active and fluid systems. Soft Matter 21, 7544-7564, 2025. DOI
From predictors and SHAKE toward unified integration scheme for molecular dynamics
Molecular dynamics (MD) simulations give us a substantially detailed insight into what is going on in a molecular system of interest. Throughout the years of their evolution, several MD algorithms have been developed. However, the initial abundant variability of integration methods for solving the underlying equations of motion has been narrowed to a very limited set of methods generally accepted as the best. Despite being appropriate in many applications, situations may occur in which alternative implementation could be advantageous. Therefore, the purpose of this work is to show an alternative universal way of MD algorithm construction based on predictors. The suggested unified integration scheme for MD includes a lot of variability that has once been lost.
To formulate such a scheme, we adapted the traditional SHAKE method for treating rigid bonds to a predictor-corrector integration scheme and combined it with the existing time-reversible velocity predictor and box predictor. This new approach enables using both the time-honored Verlet method and the recently improved Gear methods. The resulting unified integration scheme was tested on two simple models and two MD systems: SPC/E water and ionic liquid (1-ethyl-3-methylimidazolium tetrafluoroborate). Trajectories in microcanonical, Nosé–Hoover canonical, and MTK isobaric ensembles were generated. The results for Verlet-based methods were in excellent agreement with those obtained using conventional integration methods. For particular tasks, the use of higher-order methods can be beneficial. Overall, in comparison with standard approaches, our universal scheme provides a significantly simpler route to devising new integrators and maintaining existing simulation software. Furthermore, our family of new integrators can be efficiently deployed in massively parallel MD software.
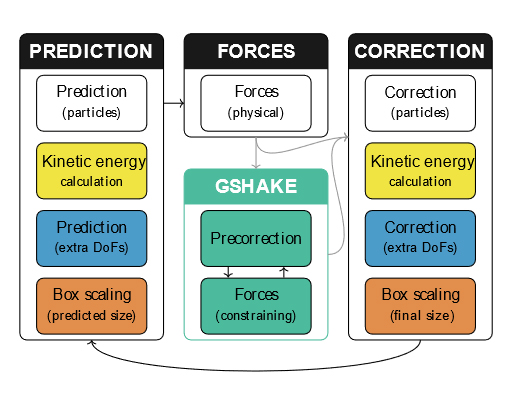
- J. Janek: From predictors and SHAKE toward unified integration scheme for molecular dynamics. J. Chem. Phys. 162, 084103, 2025. DOI
Molecular simulations of cesium halide aqueous solutions and crystalline salts using phase-transferable polarizable force fields
Cesium, an important alkali metal halide, exhibits distinct behavior necessitating accurate modeling of its interactions. We present a refined polarizable force field for Cs+ ions, extending the set of DLM/2022-BK3 FFs, to improve predictions of cesium halide properties. The refined model accurately captures structural properties, phase transitions, and concentration-dependent properties of aqueous solutions of CsF, CsCl, CsBr, and CsI, including high concentrations. Structural insights reveal variations in hydration numbers and the formation of contact ion pairs, especially pronounced with larger counterions. Notably, the force fields accurately predict aqueous solubilities, highlighting their accuracy in both ion-water and ion-ion interactions. While primarily designed for single salt solutions, these models offer broad applicability, extending to complex mixtures. Our findings underscore the reliability and versatility of the developed Cs+ model and the DLM/2022-BK3 force fields, which provides a valuable tool for studying alkali metal halide systems.
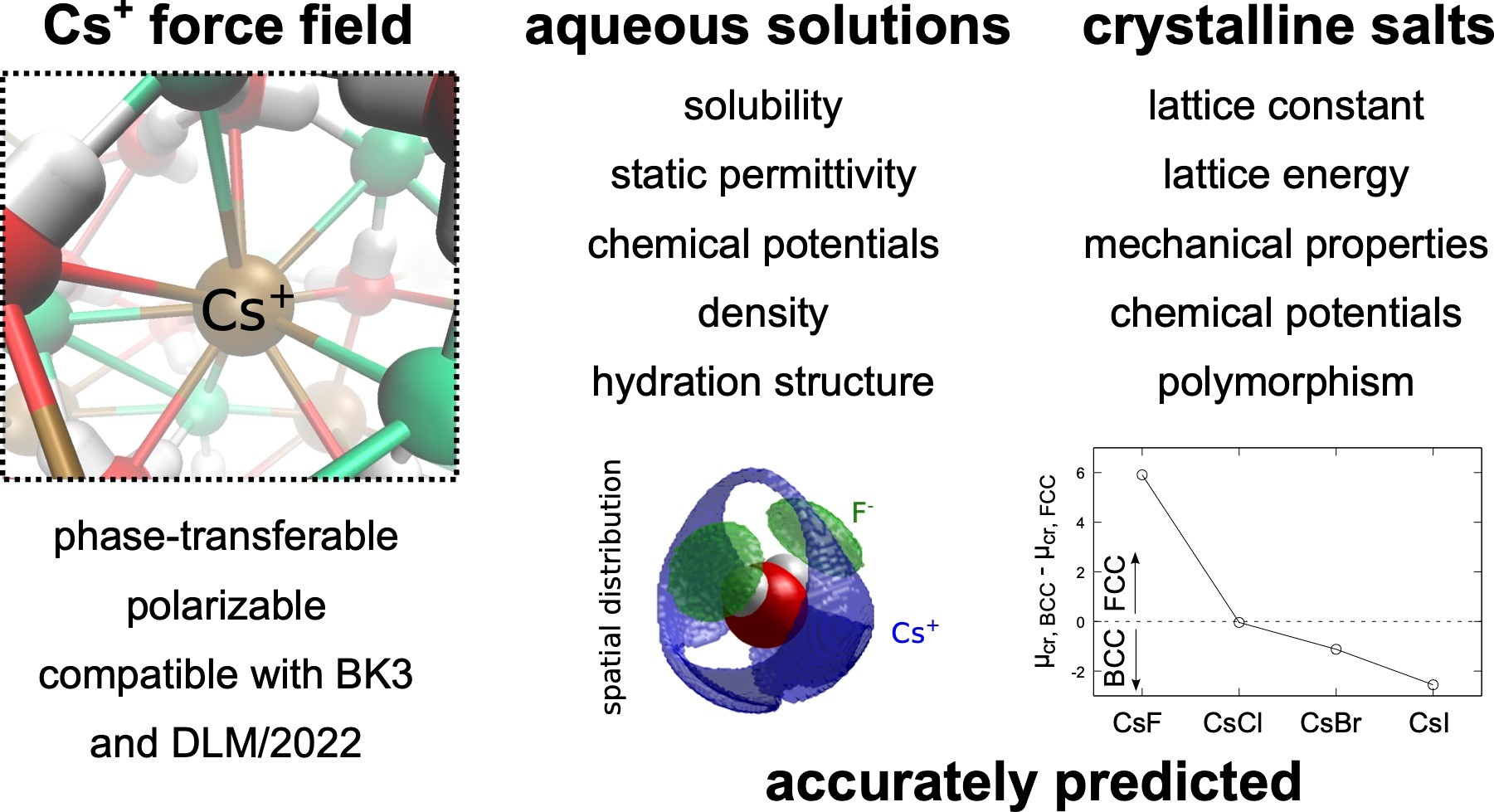
- J. Dočkal, P. Mimrová, M. Lísal, F. Moučka: Molecular simulations of cesium halide aqueous solutions and crystalline salts using phase-transferable polarizable force fields. J. Mol. Liq. 424, 127121, 2025. DOI
Understanding interfacial structure and preferential adsorption in mixed alkali-halide electrolytes at graphene oxide electrodes by constant potential molecular dynamics simulations
Graphene oxide (GO) is a promising material that finds use in electrochemical applications. Therefore, understanding the microscopic behavior of electrolytes in contact with GO electrodes is important. In this work, we focus on a detailed description and explanation of the structure, adsorption behavior, and self-diffusion of aqueous solutions of single-salt and mixed-cation alkali metal chlorides in the vicinity of hydroxylated GO electrodes under normal thermodynamic conditions and varying interelectrode voltages. We performed molecular dynamics simulations of the solutions constrained between planar GO electrodes using the constant potential method. We analyzed several structural properties, including profiles of atomic density, charge density, characteristics of the network of non-covalent bonds, and in-plane and transverse self-diffusion, all as functions of the distance from the GO surface. We discuss and explain the behavior of all these properties in detail as a result of three driving forces: (i) the direct electrode-solution interactions, (ii) the tendency of the solutions to saturate the network of non-covalent bonds, and (iii) the tendency of the system to suppress local charge accumulation in any region larger than typical interparticle distances. The existence of hydroxyl groups here greatly enhances the direct electrode-solution interactions, causing qualitative differences in the structure, including a different arrangement of the adsorption layers of ions in comparison to the solutions at graphene electrodes.
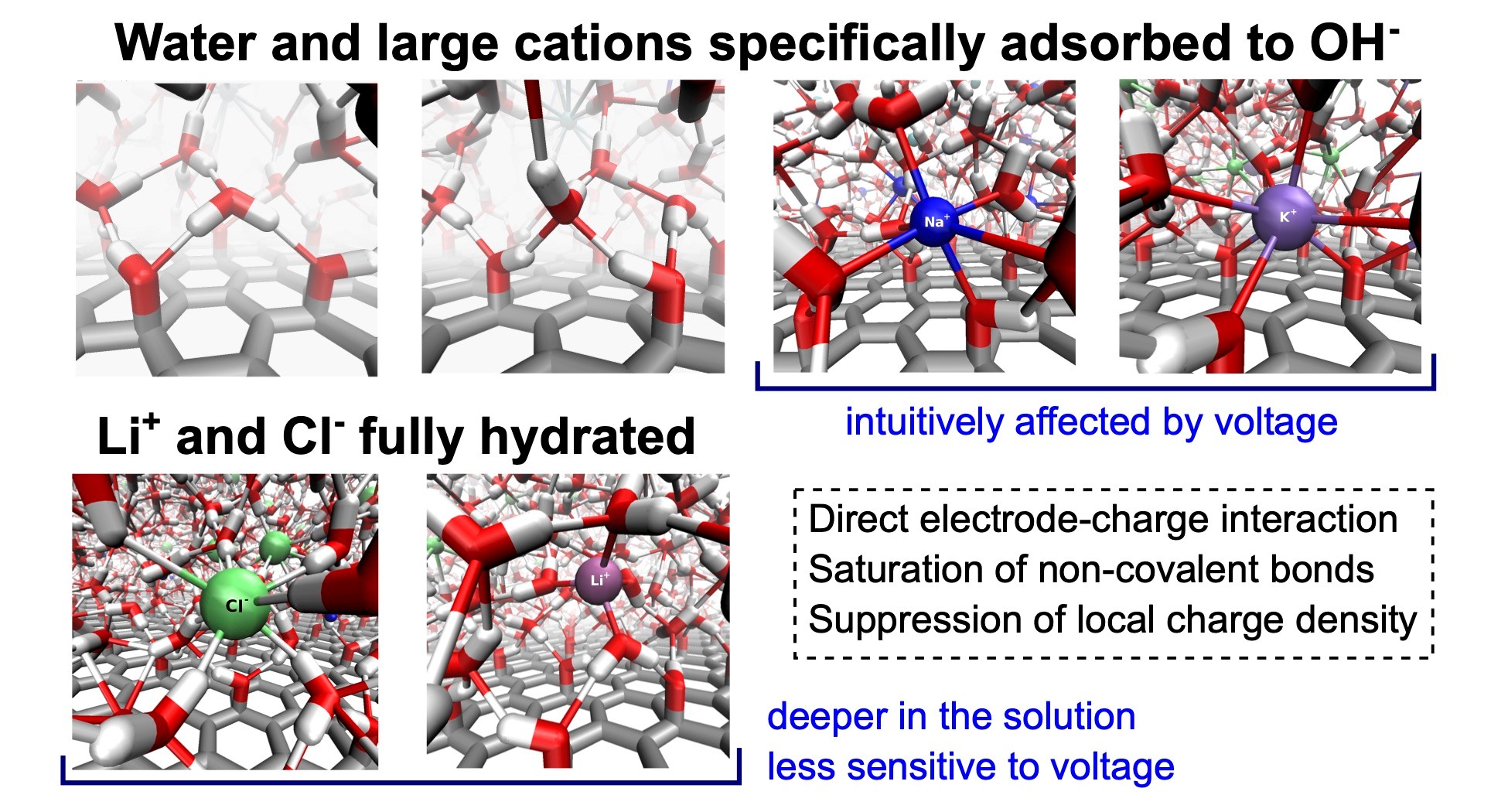
- J. Dočkal, E. Rezlerová, M. Předota, M. Lísal, F. Moučka: Understanding interfacial structure and preferential adsorption in mixed alkali-halide electrolytes at graphene oxide electrodes by constant potential molecular dynamics simulations. J. Mol. Liq. 424, 127078, 2025. DOI
Modeling temperature-dependent transport properties in dissipative particle dynamics: A top-down coarse-graining toward realistic dynamics at the mesoscale
Dissipative particle dynamics (DPD) is a widespread computational tool to simulate the behavior of soft matter and liquids in and out of equilibrium. Although there are many applications in which the effect of temperature is relevant, most of the DPD studies have been carried out at a fixed system temperature. Therefore, this work investigates how to incorporate the effect of system temperature variation within the DPD model to capture realistic temperature-dependent system properties. In particular, this work focuses on the relationship between temperature and transport properties, and therefore, an extended DPD model for transport properties prediction is employed. Transport properties, unlike the equilibrium properties, are often overlooked despite their significant influence on the flow dynamics of non-isothermal mesoscopic systems. Moreover, before simulating the response of the system induced by a temperature change, it is important to first estimate transport properties at a certain temperature. Thus here, the same fluid is simulated across different temperature conditions using isothermal DPD with the aim to identify a temperature-dependent parametrization methodology, capable of ensuring the correctness of both equilibrium and dynamical properties. Liquid water is used as a model system for these analyses. This work proposes a temperature-dependent form of the extended DPD model where both conservative and non-conservative interaction parameters incorporate the variation of the temperature. The predictions provided by our simulations are in excellent agreement with experimental data.
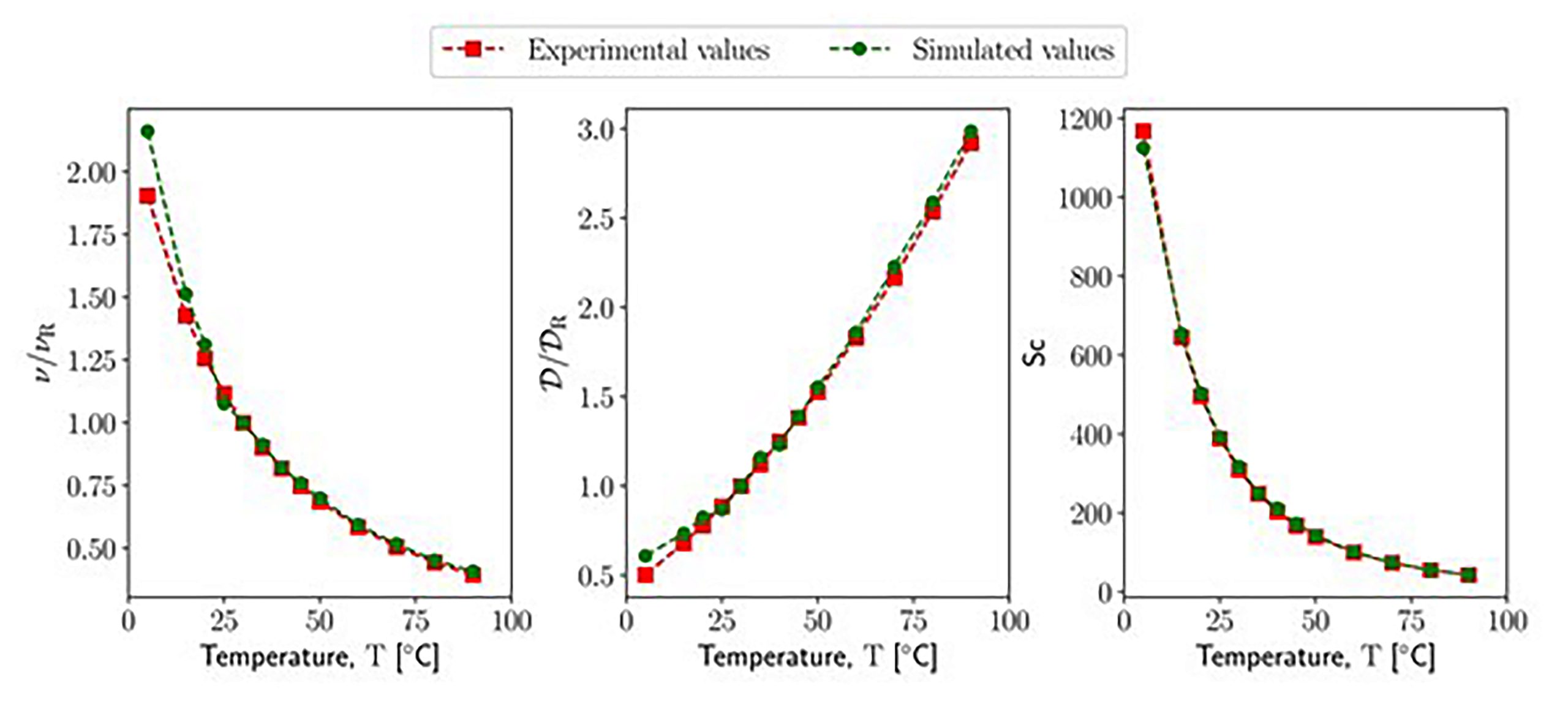
Comparison of the transport properties of water at different temperatures between experimental data and the results of DPD simulations
- N. Lauriello, M. Lísal, G. Boccardo, D. Marchisio, A. Buffo: Modeling temperature-dependent transport properties in dissipative particle dynamics: A top-down coarse-graining toward realistic dynamics at the mesoscale. J. Chem. Phys. 161(3), 034112, 2024. DOI
Generalized energy-conserving dissipative particle dynamics with mass transfer: Coupling between energy and mass exchange
The complete description of energy and material transport within the Generalized energy-conserving dissipative particle dynamics with mass transfer (GenDPDE-M) methodology is presented. In particular, the dynamic coupling between mass and energy is incorporated into the GenDPDE-M, which was previously introduced with dynamically decoupled fluxes (J. Bonet Avalos et al., J. Chem. Theory Comput. 18(12), 7639–7652, 2022). From a theoretical perspective, we have derived the appropriate Fluctuation-Dissipation theorems along with Onsager’s reciprocal relations, suitable for mesoscale models featuring this coupling. Equilibrium and non-equilibrium simulations are performed to demonstrate the internal thermodynamic consistency of the method, as well as the ability to capture the Ludwig–Soret effect and tune its strength through the mesoscopic parameters. In view of the completeness of the presented approach, GenDPDE-M is the most general Lagrangian method to deal with complex fluids and systems at the mesoscale, where thermal agitation is relevant.
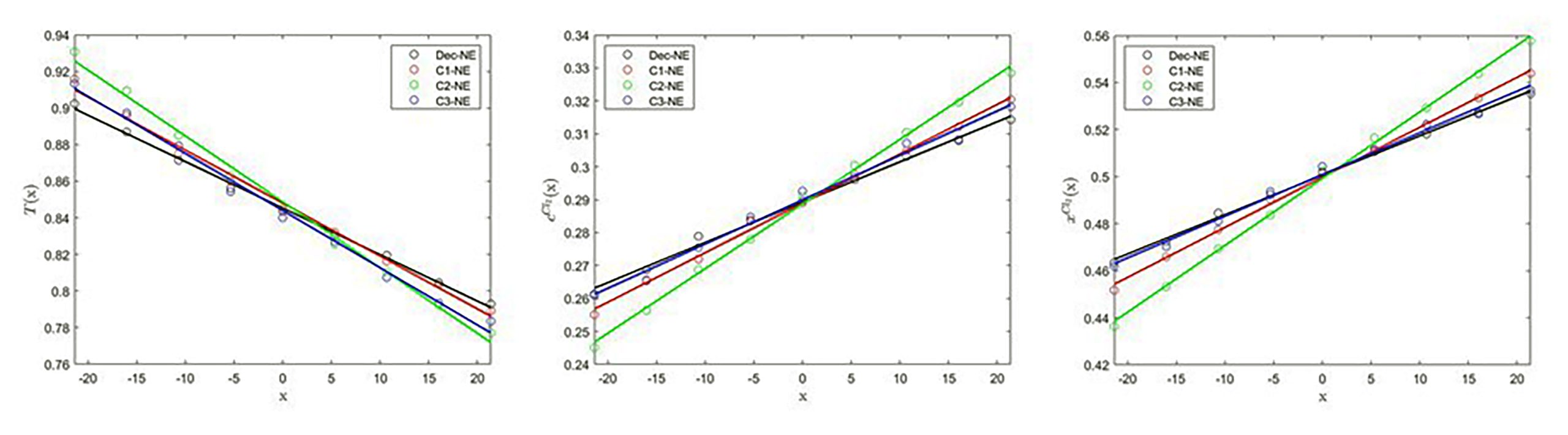
The x-profiles of the particle temperature (left), and the number concentration (middle) and molar fraction (right) of Cl2 for the coarse-grain equimolar Ar/Cl2 binary mixture
- G. Colella, A.D. Mackie, J.P. Larentzos, J.K. Brennan, M. Lísal, J. Bonet Avalos: Generalized energy-conserving dissipative particle dynamics with mass transfer: Coupling between energy and mass exchange. J. Non-Equilib. Thermodyn. 49, 347-375, 2024. DOI
Kelvin equation for bridging transitions
see Research Achievements of the Institute
- A. Malijevský, M. Pospíšil: Kelvin equation for bridging transitions. Phys. Rev. E 109(3 March), 034801, 2024. DOI
Capillary condensation between nonparallel walls
see Research Achievements of the Institute.
- A. Malijevský, J. Janek: Capillary condensation between nonparallel walls. Phys. Rev. E 109, 054801, 2024. DOI
Structure and self-diffusivity of mixed-cation electrolytes between neutral and charged graphene sheets
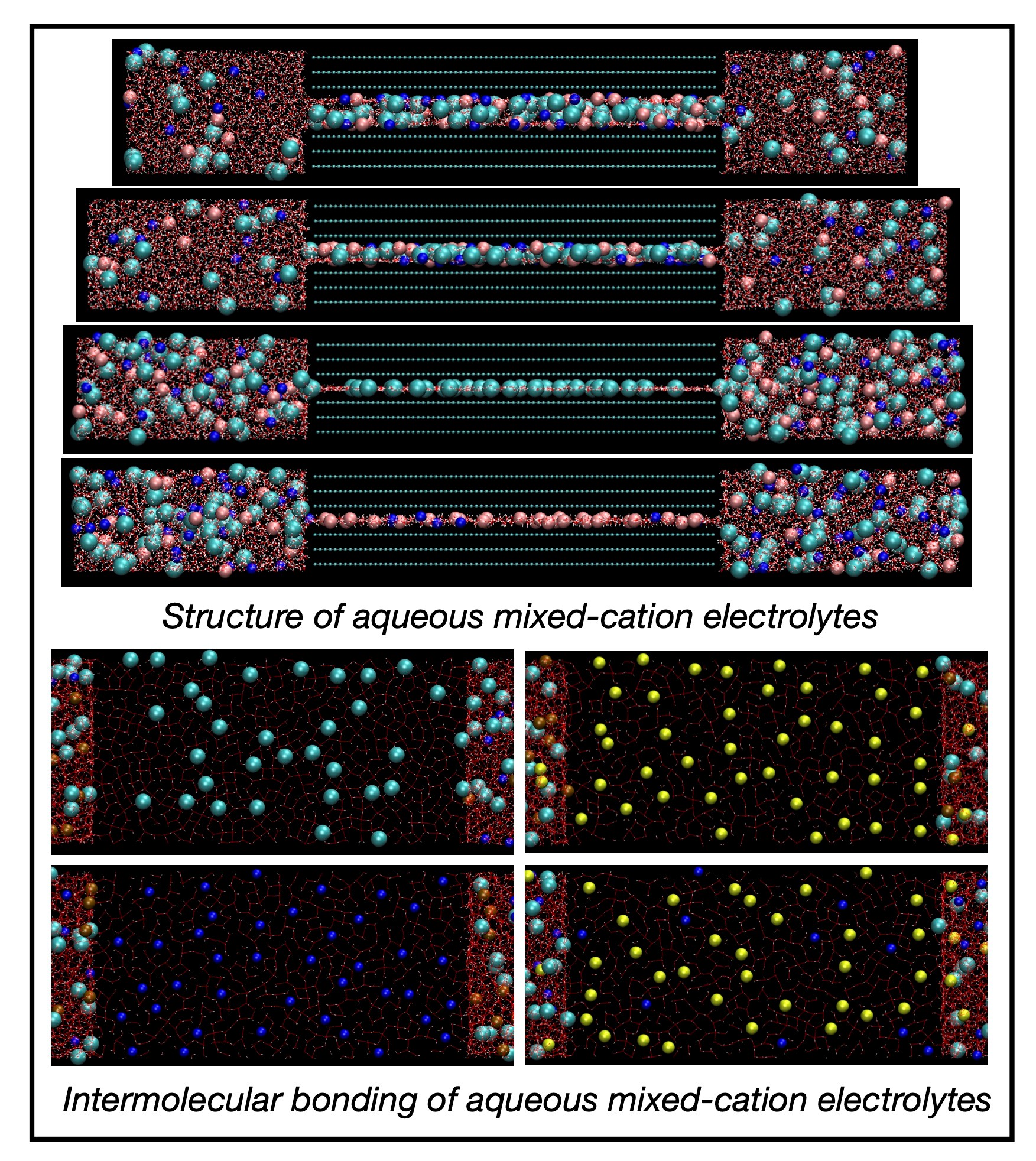 Graphene-based applications, such as supercapacitors or capacitive deionization, take place in an aqueous environment, and they benefit from molecular-level insights into the behavior of aqueous electrolyte solutions in single-digit graphene nanopores with a size comparable to a few molecular diameters. Under single-digit graphene nanoconfinement (smallest dimension < 2 nm), water and ions behave drastically different than in the bulk. Most aqueous electrolytes in graphene-based applications as well as in nature contain a mix of electrolytes. We study several prototypical aqueous mixed alkali-chloride electrolytes containing an equimolar fraction of Li/Na, Li/K, or Na/K cations confined between neutral and positively or negatively charged parallel graphene sheets. The strong hydration shell of small Li+ vs a larger Na+ or large K+ with weaker or weak hydration shells affects the interplay between the ions’ propensity to hydrate or dehydrate under the graphene nanoconfinement and the strength of the ion–graphene interactions mediated by confinement-induced layered water. We perform molecular dynamics simulations of the confined mixed-cation electrolytes using the effectively polarizable force field for electrolyte–graphene systems and focus on a relation between the electrochemical adsorption and structural properties of the water molecules and ions and their diffusion behavior. The simulations show that the one-layer nanoslits have the biggest impact on the ions’ adsorption and the water and ions’ diffusion. The positively charged one-layer nanoslits only allow for Cl− adsorption and strengthen the intermolecular bonding, which along with the ultrathin confinement substantially reduces the water and Cl− diffusion. In contrast, the negatively charged one-layer nanoslits only allow for the adsorption of weakly hydrated Na+ or K+ and substantially break up the non-covalent bond network, which leads to the enhancement of the water and Na+ or K+ diffusion up to or even above the bulk diffusion. In wider nanoslits, cations adsorb closer to the graphene surfaces than Cl−’s with preferential adsorption of a weakly hydrated cation over a strongly hydrated cation. The positive graphene charge has an intuitive effect on the adsorption of weakly hydrated Na+’s or K+’s and Cl−’s and a counterintuitive effect on the adsorption of strongly hydrated Li+’s. On the other hand, the negative surface charge has an intuitive effect on the adsorption of both types of cations and only mild intuitive or counterintuitive effects on the Cl− adsorption. The diffusion of water molecules and ions confined in the wider nanoslits is reduced with respect to the bulk diffusion, more for the positive graphene charge, which strengthened the intermolecular bonding, and less for the negative surface charge, which weakened the non-covalent bond network.
Graphene-based applications, such as supercapacitors or capacitive deionization, take place in an aqueous environment, and they benefit from molecular-level insights into the behavior of aqueous electrolyte solutions in single-digit graphene nanopores with a size comparable to a few molecular diameters. Under single-digit graphene nanoconfinement (smallest dimension < 2 nm), water and ions behave drastically different than in the bulk. Most aqueous electrolytes in graphene-based applications as well as in nature contain a mix of electrolytes. We study several prototypical aqueous mixed alkali-chloride electrolytes containing an equimolar fraction of Li/Na, Li/K, or Na/K cations confined between neutral and positively or negatively charged parallel graphene sheets. The strong hydration shell of small Li+ vs a larger Na+ or large K+ with weaker or weak hydration shells affects the interplay between the ions’ propensity to hydrate or dehydrate under the graphene nanoconfinement and the strength of the ion–graphene interactions mediated by confinement-induced layered water. We perform molecular dynamics simulations of the confined mixed-cation electrolytes using the effectively polarizable force field for electrolyte–graphene systems and focus on a relation between the electrochemical adsorption and structural properties of the water molecules and ions and their diffusion behavior. The simulations show that the one-layer nanoslits have the biggest impact on the ions’ adsorption and the water and ions’ diffusion. The positively charged one-layer nanoslits only allow for Cl− adsorption and strengthen the intermolecular bonding, which along with the ultrathin confinement substantially reduces the water and Cl− diffusion. In contrast, the negatively charged one-layer nanoslits only allow for the adsorption of weakly hydrated Na+ or K+ and substantially break up the non-covalent bond network, which leads to the enhancement of the water and Na+ or K+ diffusion up to or even above the bulk diffusion. In wider nanoslits, cations adsorb closer to the graphene surfaces than Cl−’s with preferential adsorption of a weakly hydrated cation over a strongly hydrated cation. The positive graphene charge has an intuitive effect on the adsorption of weakly hydrated Na+’s or K+’s and Cl−’s and a counterintuitive effect on the adsorption of strongly hydrated Li+’s. On the other hand, the negative surface charge has an intuitive effect on the adsorption of both types of cations and only mild intuitive or counterintuitive effects on the Cl− adsorption. The diffusion of water molecules and ions confined in the wider nanoslits is reduced with respect to the bulk diffusion, more for the positive graphene charge, which strengthened the intermolecular bonding, and less for the negative surface charge, which weakened the non-covalent bond network.
- E. Rezlerová, F. Moučka, M. Předota, M. Lísal: Structure and self-diffusivity of mixed-cation electrolytes between neutral and charged graphene sheets, J. Chem. Phys. 160, 094701, 2024. DOI
Green-Kubo expressions for transport coefficients from dissipative particle dynamics simulations revisited
 This article addresses the debate about the correct application of Green–Kubo expressions for transport coefficients from dissipative particle dynamics simulations. We demonstrate that the Green–Kubo expressions are valid provided that (i) the dynamic model conserves the physical property, whose transport is studied, and (ii) the fluctuations satisfy detailed balance. As a result, the traditional expressions used in molecular dynamics can also be applied to dissipative particle dynamics simulations. However, taking the calculation of the shear viscosity as a paradigmatic example, a random contribution, whose strength scales as 1/dt1/2, with dt the timestep, can cause difficulties if the stress tensor is not separated into the different contributions. We compare our expression to that of Ernst and Brito (M. H. Ernst and R. Brito, Europhys. Lett. 73, 183-189, 2006), which arises from a diametrically different perspective. We demonstrate that the two expressions are completely equivalent and find exactly the same result both analytically and numerically. We show that the differences are not due to the lack of time-reversibility but instead from a preaveraging of the random contributions. Despite the overall validity of Green–Kubo expressions, we find that the Einstein–Helfand relations (D. C. Malaspina et al. Phys. Chem. Chem. Phys. 25, 12025-12040, 2023) do not suffer from the need to decompose the stress tensor and can readily be used with a high degree of accuracy. Consequently, Einstein–Helfand relations should be seen as the preferred method to calculate transport coefficients from dissipative particle dynamics simulations.
This article addresses the debate about the correct application of Green–Kubo expressions for transport coefficients from dissipative particle dynamics simulations. We demonstrate that the Green–Kubo expressions are valid provided that (i) the dynamic model conserves the physical property, whose transport is studied, and (ii) the fluctuations satisfy detailed balance. As a result, the traditional expressions used in molecular dynamics can also be applied to dissipative particle dynamics simulations. However, taking the calculation of the shear viscosity as a paradigmatic example, a random contribution, whose strength scales as 1/dt1/2, with dt the timestep, can cause difficulties if the stress tensor is not separated into the different contributions. We compare our expression to that of Ernst and Brito (M. H. Ernst and R. Brito, Europhys. Lett. 73, 183-189, 2006), which arises from a diametrically different perspective. We demonstrate that the two expressions are completely equivalent and find exactly the same result both analytically and numerically. We show that the differences are not due to the lack of time-reversibility but instead from a preaveraging of the random contributions. Despite the overall validity of Green–Kubo expressions, we find that the Einstein–Helfand relations (D. C. Malaspina et al. Phys. Chem. Chem. Phys. 25, 12025-12040, 2023) do not suffer from the need to decompose the stress tensor and can readily be used with a high degree of accuracy. Consequently, Einstein–Helfand relations should be seen as the preferred method to calculate transport coefficients from dissipative particle dynamics simulations.
- D. C. Malaspina, M. Lísal, J. P. Larentzos, J. K. Brennan, A. D. Mackie, J. Bonet Avalos, Green-Kubo expressions for transport coefficients from dissipative particle dynamics simulations revisited. Phys. Chem. Chem. Phys. 26, 1328-1339, 2024. DOI
Structure of aqueous alkali metal halide electrolyte solutions from molecular simulations of phase-transferable polarizable models
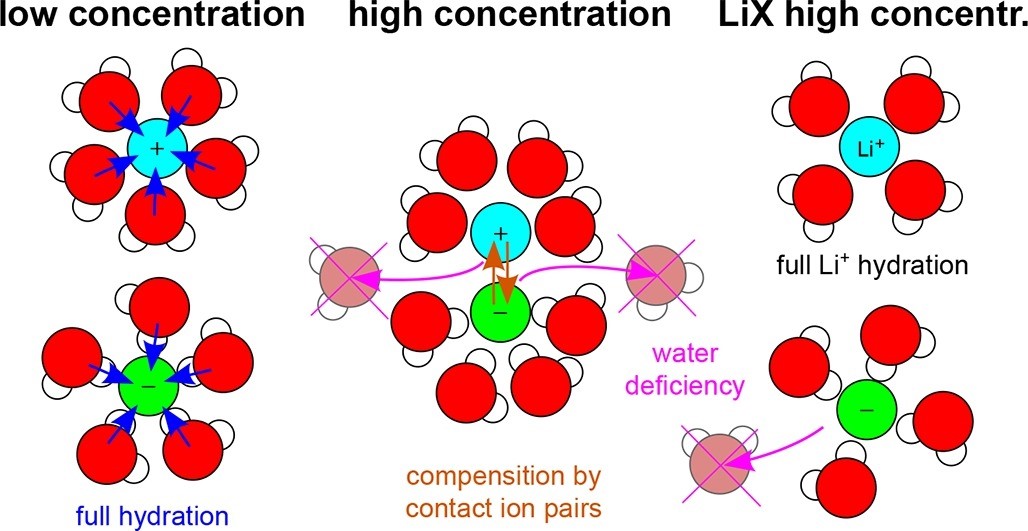 The structure of aqueous solutions of alkali metal halides is studied under ambient thermodynamic conditions and concentrations from infinite dilution to supersaturation using molecular dynamics simulations of phase-transferable polarizable models. The results of the solution densities, radial distribution functions, 3D spatial distribution functions, the properties of hydrogen and other noncovalent bonds in solutions, hydration numbers, coordination numbers, numbers of contact cation-anion pairs, and other statistics of the number of ions hydrated simultaneously by a shared water molecule are systematically presented. In particular, the results show and quantify how the strengths of the hydration bonds of different ions vary and how the hydration numbers decrease with increasing concentration in parallel with an increase in the number of contact cation-anion pairs. In most cases, they completely compensate for the loss of water-ion bonds by an increase in cation-anion bonds. The exceptions are solutions based on the Li+ cation, which retain a solid hydration shell even at high concentrations. This behavior is conceptualized based on three imaginary driving forces: the first dominating at low concentrations and causing full hydration of the ions, the second representing a lack of water necessary for full hydration of the ions and increasing with increasing concentration, and the third attracting counterions to the water-unoccupied sites of the hydration shells and also increasing with concentration. This concept can be used not only to understand the structural behavior of homogeneous electrolytes in thermodynamic equilibrium but also to study phenomena that involve preferential adsorption of ions on electrodes, nanochannels, or porous materials. The data obtained for the number and strength of hydration bonds and ion pairs can also be used in further studies to elucidate the diffusion behavior, viscosity, and conductivity of aqueous electrolyte solutions.
The structure of aqueous solutions of alkali metal halides is studied under ambient thermodynamic conditions and concentrations from infinite dilution to supersaturation using molecular dynamics simulations of phase-transferable polarizable models. The results of the solution densities, radial distribution functions, 3D spatial distribution functions, the properties of hydrogen and other noncovalent bonds in solutions, hydration numbers, coordination numbers, numbers of contact cation-anion pairs, and other statistics of the number of ions hydrated simultaneously by a shared water molecule are systematically presented. In particular, the results show and quantify how the strengths of the hydration bonds of different ions vary and how the hydration numbers decrease with increasing concentration in parallel with an increase in the number of contact cation-anion pairs. In most cases, they completely compensate for the loss of water-ion bonds by an increase in cation-anion bonds. The exceptions are solutions based on the Li+ cation, which retain a solid hydration shell even at high concentrations. This behavior is conceptualized based on three imaginary driving forces: the first dominating at low concentrations and causing full hydration of the ions, the second representing a lack of water necessary for full hydration of the ions and increasing with increasing concentration, and the third attracting counterions to the water-unoccupied sites of the hydration shells and also increasing with concentration. This concept can be used not only to understand the structural behavior of homogeneous electrolytes in thermodynamic equilibrium but also to study phenomena that involve preferential adsorption of ions on electrodes, nanochannels, or porous materials. The data obtained for the number and strength of hydration bonds and ion pairs can also be used in further studies to elucidate the diffusion behavior, viscosity, and conductivity of aqueous electrolyte solutions.
- J. Dočkal, P. Mimrová, M. Lísal, F. Moučka, Structure of aqueous alkali metal halide electrolyte solutions from molecular simulations of phase-transferable polarizable models. J. Mol. Liq. 394, 123797, 2024. DOI
Structure and self-diffusivity of alkali-halide electrolytes in neutral and charged graphene nanochannels
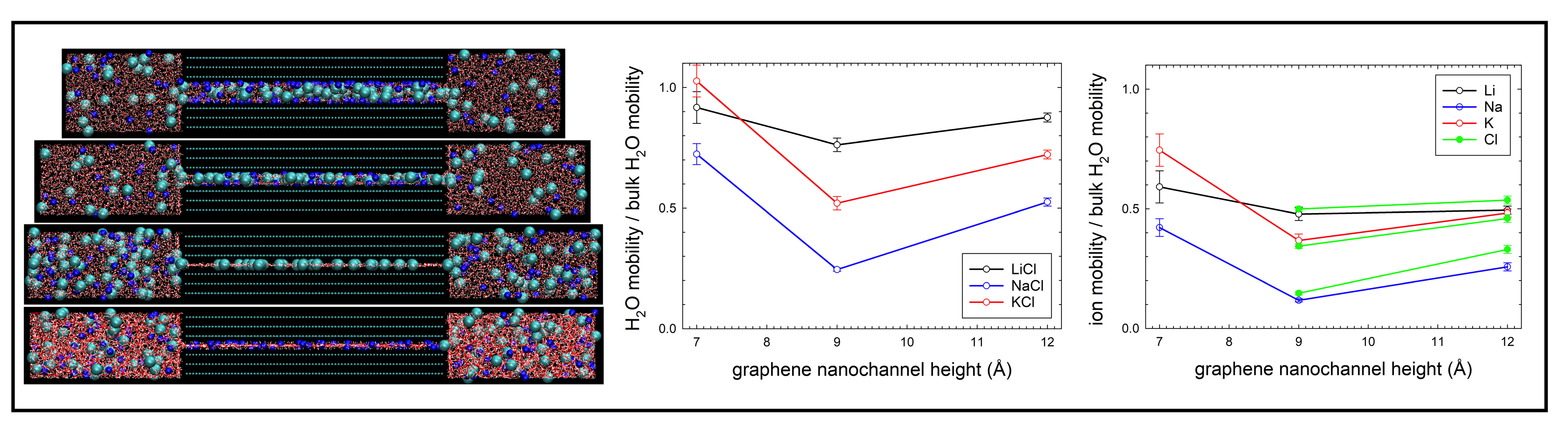 Understanding the microscopic behavior of aqueous electrolyte solutions in graphene-based ultrathin nanochannels is important in nanofluidic applications such as water purification, fuel cells, and molecular sensing. Under extreme confinement (< 2 nm), the properties of water and ions differ drastically from those in the bulk phase. We studied the structural and diffusion behavior of prototypical aqueous solutions of electrolytes (LiCl, NaCl, and KCl) confined in both neutral and positively-, and negatively-charged graphene nanochannels. We performed molecular dynamics simulations of the solutions in the nanochannels with either one, two- or three-layer water structures using the effectively polarisable force field for graphene.
Understanding the microscopic behavior of aqueous electrolyte solutions in graphene-based ultrathin nanochannels is important in nanofluidic applications such as water purification, fuel cells, and molecular sensing. Under extreme confinement (< 2 nm), the properties of water and ions differ drastically from those in the bulk phase. We studied the structural and diffusion behavior of prototypical aqueous solutions of electrolytes (LiCl, NaCl, and KCl) confined in both neutral and positively-, and negatively-charged graphene nanochannels. We performed molecular dynamics simulations of the solutions in the nanochannels with either one, two- or three-layer water structures using the effectively polarisable force field for graphene.
We analyzed the structure and intermolecular bond network of the confined solutions along with their relation to the self-diffusivity of water and ions. The simulations show that Na and K cations can more easily rearrange their solvation shells under the graphene nanoconfinement and adsorb on the graphene surfaces or dissolve in the confinement-induced layered water than the Li cation. The negative surface charge together with the presence of ions orient water molecules with hydrogens towards the graphene surfaces, which in turn weakens the intermolecular bond network. The one-layer nanochannels have the biggest effect on the water structure and intermolecular bonding as well as on the adsorption of ions with only co-ions entering these nanochannels. The self-diffusivity of confined water is strongly reduced with respect to the bulk water and decreases with diminishing nanochannel heights except for the negatively-charged one-layer nanochannel. The self-diffusivity of ions also decreases with the reducing the nanochannel heights except for the self-diffusivity of cations in the negatively-charged one-layer nanochannel, evidencing cooperative diffusion of confined water and ions. Due to the significant break-up of the intermolecular bond network in the negatively-charged one-layer nanochannel, self-diffusion coefficients of water and cations exceed those for the two- and three-layer nanochannels and become comparable to the bulk values.
- E. Rezlerová, F. Moučka, M. Předota, M. Lísal, Structure and self-diffusivity of alkali-halide electrolytes in neutral and charged graphene nanochannels, Phys. Chem. Chem. Phys. 25, 21579-21594, 2023. DOI
Development of an automated reliable method to compute transport properties from DPD equilibrium simulations: Application to simple fluids
 Dissipative particle dynamics (DPD) is a promising candidate technique for modeling the rheological properties of soft matter systems. However, several methodological issues inhibit its exploitation as a computational rheology tool. In this work, we focus on the development of an automated method to compute transport properties from equilibrium simulation with particular attention to the assessment of the Green-Kubo approach reliability and computational feasibility for a large set of simple DPD systems with increasing Schmidt number. Furthermore, we investigate the time step size dependency of dynamic properties and the role of different time integration schemes. In particular, we assess the performance of the Shardlow-splitting algorithm against the most popular modified velocity-Verlet algorithm. We consider, for the first time, the application of the Shardlow-splitting algorithm to the transverse DPD thermostat in different friction regimes relying on systematic numerical experiments. In addition, we make use of these findings to perform a multi-parametric study aiming to investigate the Schmidt number relationship with the effective friction coefficient.
Dissipative particle dynamics (DPD) is a promising candidate technique for modeling the rheological properties of soft matter systems. However, several methodological issues inhibit its exploitation as a computational rheology tool. In this work, we focus on the development of an automated method to compute transport properties from equilibrium simulation with particular attention to the assessment of the Green-Kubo approach reliability and computational feasibility for a large set of simple DPD systems with increasing Schmidt number. Furthermore, we investigate the time step size dependency of dynamic properties and the role of different time integration schemes. In particular, we assess the performance of the Shardlow-splitting algorithm against the most popular modified velocity-Verlet algorithm. We consider, for the first time, the application of the Shardlow-splitting algorithm to the transverse DPD thermostat in different friction regimes relying on systematic numerical experiments. In addition, we make use of these findings to perform a multi-parametric study aiming to investigate the Schmidt number relationship with the effective friction coefficient.
- N. Lauriello, G. Boccardo, D. Marchisio, M. Lísal, A. Buffo, Development of an automated reliable method to compute transport properties from DPD equilibrium simulations: Application to simple fluids, Comput. Phys. Commun. 291, 108843, 2023. DOI
Interactions of cationic surfactant-fatty alcohol monolayers with natural human hair surface: Insights from dissipative particle dynamics
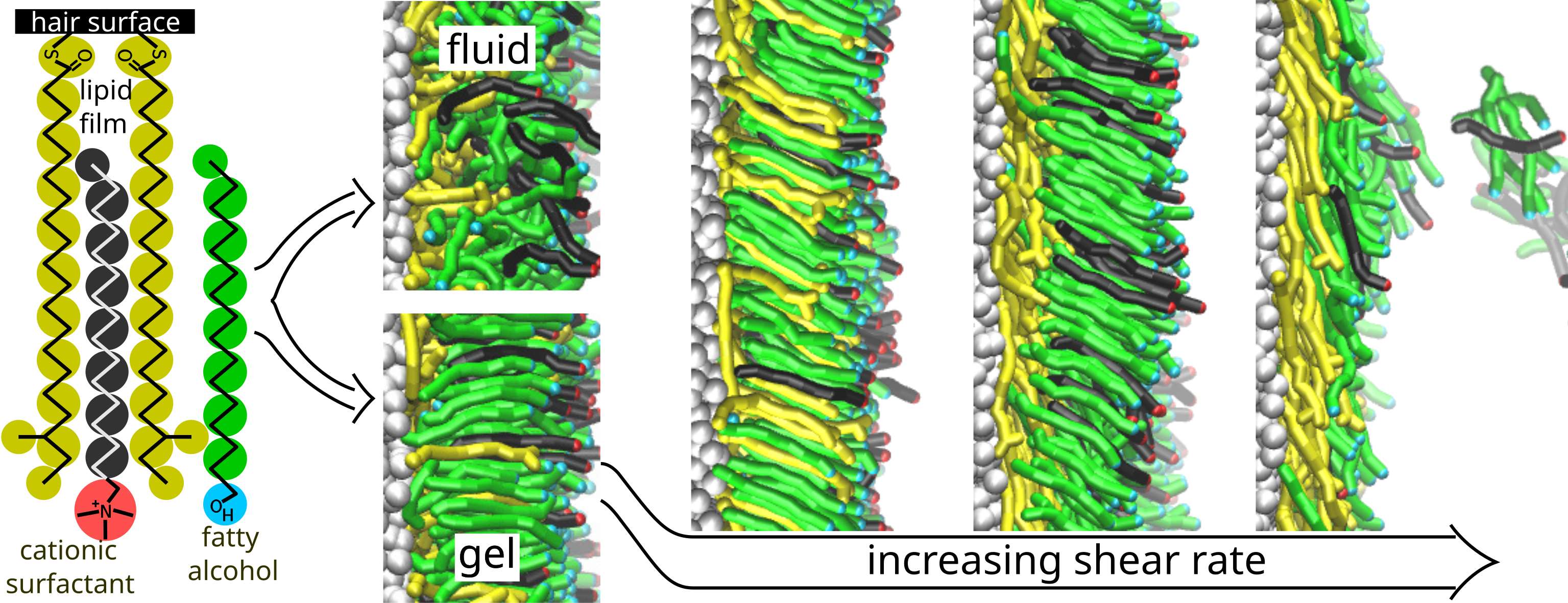 Fatty alcohols (CnFAs) combined with cationic surfactants are common ingredients of lamellar-phase personal care liquids. We employ mesoscopic modelling to study how surfactant-CnFA monolayers originating from the corresponding bilayers of the personal care liquids interact with the human hair surface above and below the fluid-gel transition temperature as well as in- and out-of-equilibrium. For the mono-layer model, we consider the single-tail cationic surfactant cetyltrimethylammonium chloride (CTAC) and an excess of CnFAs with their alkyl tail length equal to or longer than the CTAC alkyl tail length.
Fatty alcohols (CnFAs) combined with cationic surfactants are common ingredients of lamellar-phase personal care liquids. We employ mesoscopic modelling to study how surfactant-CnFA monolayers originating from the corresponding bilayers of the personal care liquids interact with the human hair surface above and below the fluid-gel transition temperature as well as in- and out-of-equilibrium. For the mono-layer model, we consider the single-tail cationic surfactant cetyltrimethylammonium chloride (CTAC) and an excess of CnFAs with their alkyl tail length equal to or longer than the CTAC alkyl tail length.
The hair surface mimics keratin surface proteins covered by a film of lipid chains covalently bonded to the proteins. Our modelling shows the formation of a dense adsorbed layer due to the interactions of the CTAC and CnFA alkyl tails with the hydrophobic hair surface. The adsorption and the behaviour of the adsorbed layer is different under fluid and gel conditions. The differences are related to the structure of the adsorbed layer as characterised by density profiles across the adsorbed layer and the orientational order parameters of the chains within the adsorbed layer. Under steady-state shearing (an approximation of real, non-equilibrium conditions), increasing the shear rate above a threshold leads to continuous or abrupt desorption of the CTAC and CnFA chains under fluid or gel conditions, respectively; the desorbed chains can then form various self-assembled structures in the bulk solution. The underlying mechanism of CTAC and CnFA desorption from the adsorbed layers is closely related to the corresponding adsorption mechanism.
- K. Šindelka, A. Kowalski, M. Cooke, C. Mendoza, M. Lísal, Interactions of cationic surfactant-fatty alcohol monolayers with natural human hair surface: Insights from dissipative particle dynamics, J. Mol. Liq. 375, 121385, 2023. DOI
Pore pressure drop during dynamic rupture and conditions for dilatancy hardening
Cores of geological faults are granular layers often filled with fluid, which tend to dilate on increase in slip rate, for example, during an earthquake nucleation. Dilation of the fault causes a drop in pore fluid pressure which in turn increases strength of the fault. This effect, known as dilatancy hardening, may suppress generation of earthquakes. However, the magnitude of fluid depressurization during dynamic slip is difficult to measure. We systematically study pore pressure drop following the onset of slip in fluid-saturated granular layers, using theory and numerical simulations. We find two regimes with distinct evolutions of pore pressure and dilatation. For slow slip rate, high permeability or high stress acting upon the layer (drained conditions), the pore pressure drop is quickly restored and its ability to suppress earthquake nucleation is weak. Faster slip rate, lower permeability or stress (undrained conditions) lead to an enduring pore pressure drop, which significantly increases strength of the layer. The growing magnitude of the pore pressure drop with slip rate promotes the stability of shear rupture. Our results can be used to better constrain drainage conditions associated with changes in slip rate, the magnitude of the generated pore pressure and the corresponding fault strengthening.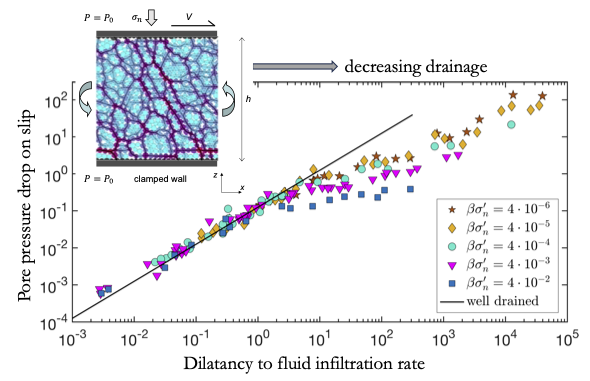
- Parez, S., Kozakovic, M., and Havlica, J., Pore pressure drop during dynamic rupture and conditions for dilatancy hardening. Journal of Geophysical Research: Solid Earth 128, e2023JB026396, 2023. https://doi.org/10.1029/2023JB026396
Recognition of the granular airborne portion in a flighted rotary drum
This ar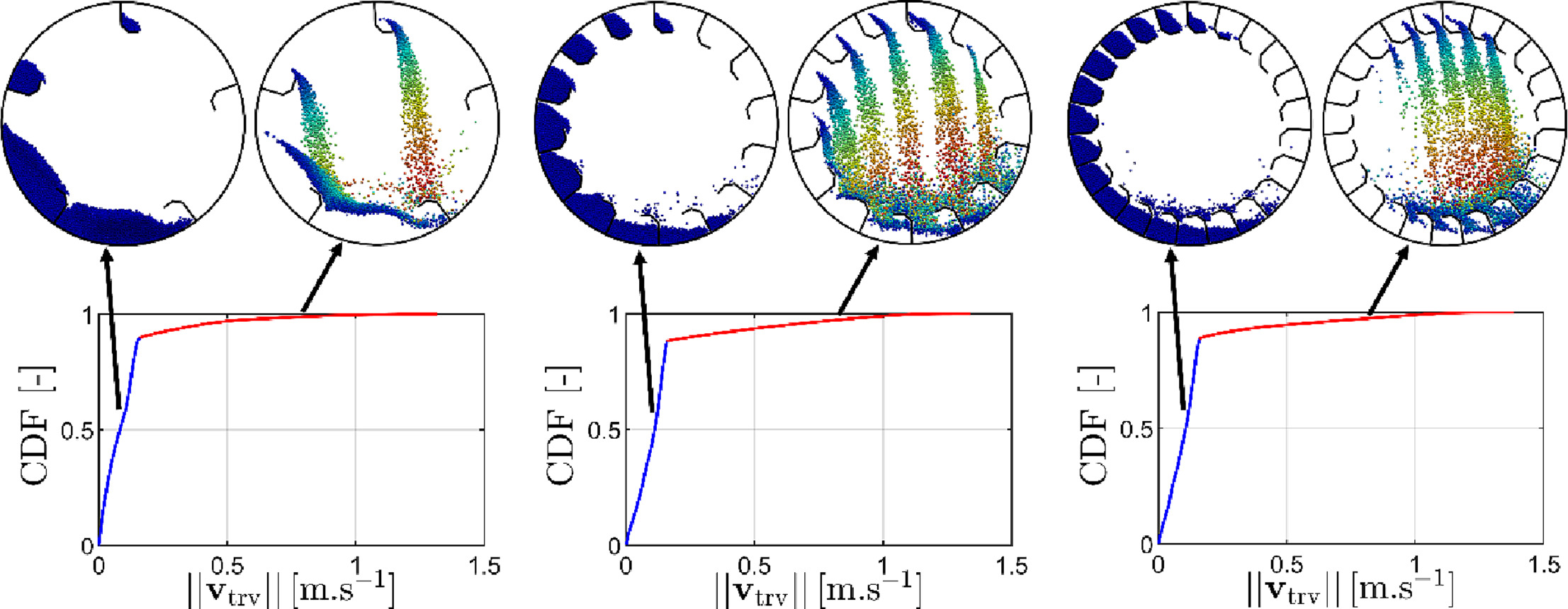 ticle deals with numerical simulation via Discrete Element Method (DEM) in a flighted rotary drum with a varying number of flights from 1 to 25 at a constant load of particles. The granular kinematics is studied in the three regimes: under-, design- and over-loading. It was shown that the behavior of granular material during the unloading process from the flights governs the regime transitions. The cross-sectional distribution analysis of the granular material in the different parts of the drum established the existence of two dense media (bed and flights) and one dilute medium (in the airborne portion). Since one of the criteria that affect the efficiency of heat transport between the gas and the particle media is the number of particles in the airborne phase, three recognition methods are proposed to detect the airborne portion. Based on geometry, velocity magnitude, and minimal separation distance, the comparison between them provides a basis for discussion about the relevance of each method in detecting airborne particles.
ticle deals with numerical simulation via Discrete Element Method (DEM) in a flighted rotary drum with a varying number of flights from 1 to 25 at a constant load of particles. The granular kinematics is studied in the three regimes: under-, design- and over-loading. It was shown that the behavior of granular material during the unloading process from the flights governs the regime transitions. The cross-sectional distribution analysis of the granular material in the different parts of the drum established the existence of two dense media (bed and flights) and one dilute medium (in the airborne portion). Since one of the criteria that affect the efficiency of heat transport between the gas and the particle media is the number of particles in the airborne phase, three recognition methods are proposed to detect the airborne portion. Based on geometry, velocity magnitude, and minimal separation distance, the comparison between them provides a basis for discussion about the relevance of each method in detecting airborne particles.
- M. Kozakovic, J. Havlica, L. Le Guen, S. Parez, F. Huchet, Recognition of the granular airborne portion in a flighted rotary drum, Powder Technology 425, 118565, 2023. DOI
Phase transitions and droplet shapes above a wetting stripe
We consider fluid adsorption on a planar wall patterned by a single stripe of width L of a wet material embedded within a partially wet surface. If L were infinite we suppose that the wall would exhibit a first-order wetting transition at temperature Tw, and an associated line of pre-wetting transitions extending off bulk-coexistence. For finite widths L, we a) determine the finite-size scaling shift of the wetting temperature Tw(L), b) derive an equation, analogous to the Kelvin equation for capillary condensation, for the shift of the pre-wetting line but involving boundary tensions, and a boundary contact angle and c) highlight the scaling and conformally invariant properties of condensed liquid drops lying above the stripe at bulk coexistence. We point out analogies between these predictions and those for capillary condensation and criticality in a parallel plate geometry and test them against numerical results obtained from a microscopic non-local classical density functional theory.
- M. Pospíšil, A. O. Parry, A. Malijevský, Phase transitions and droplet shapes above a wetting stripe, J. Mol. Liq. 381, 121834, 2023. DOI
Molecular simulations of alkali metal halide hydrates
 It has been known that classical molecular simulations of concentrated aqueous electrolyte solutions are limited by the ability of microscopic models (force fields) to predict the correct values of salt solubility, which also requires their ability to faithfully model crystalline salts. Previous simulation studies have often focused on the solubility of anhydrous crystalline salts, but virtually never on crystalline hydrates, except for hydrohalite, NaCl.2H2O, despite there are at least 23 experimentally known different hydrates that can precipitate from alkali-halide solutions. This work attempts to fill this gap in hydrate simulation studies by systematically investigating the ability of the best force fields selected to qualitatively capture the stability of the individual phases of various alkali-halide hydrates and to quantitatively predict their lattice parameters. First, we show that the nonpolarizable force fields studied often fail to model hydrates containing the Li+ cations, whereas the polarizable force fields recently refined by us are able to model all the hydrates except for LiCl.H2O. Second, we further refine our FFs for Li+ to yield stable LiCl.H2O. Third, our simulations clarify the positions of the Li+ cations in the phases of LiBr.H2O and LiI.H2O, whose distributions were previously described only as stochastic. As a byproduct, a simple and reliable simulation methodology suitable also for complex polarizable models and nonorthorhombic crystal lattices is proposed and tested, based on simulations of finite crystals floating in a vacuum.
It has been known that classical molecular simulations of concentrated aqueous electrolyte solutions are limited by the ability of microscopic models (force fields) to predict the correct values of salt solubility, which also requires their ability to faithfully model crystalline salts. Previous simulation studies have often focused on the solubility of anhydrous crystalline salts, but virtually never on crystalline hydrates, except for hydrohalite, NaCl.2H2O, despite there are at least 23 experimentally known different hydrates that can precipitate from alkali-halide solutions. This work attempts to fill this gap in hydrate simulation studies by systematically investigating the ability of the best force fields selected to qualitatively capture the stability of the individual phases of various alkali-halide hydrates and to quantitatively predict their lattice parameters. First, we show that the nonpolarizable force fields studied often fail to model hydrates containing the Li+ cations, whereas the polarizable force fields recently refined by us are able to model all the hydrates except for LiCl.H2O. Second, we further refine our FFs for Li+ to yield stable LiCl.H2O. Third, our simulations clarify the positions of the Li+ cations in the phases of LiBr.H2O and LiI.H2O, whose distributions were previously described only as stochastic. As a byproduct, a simple and reliable simulation methodology suitable also for complex polarizable models and nonorthorhombic crystal lattices is proposed and tested, based on simulations of finite crystals floating in a vacuum.
- P. Matysová, M. Lísal, F. Moučka, Molecular simulations of alkali metal halide hydrates, J. Mol. Liq. 384, 122197, 2023. DOI
Transport coefficients from Einstein-Helfand relations using standard and energy-conserving dissipative particle dynamics methods
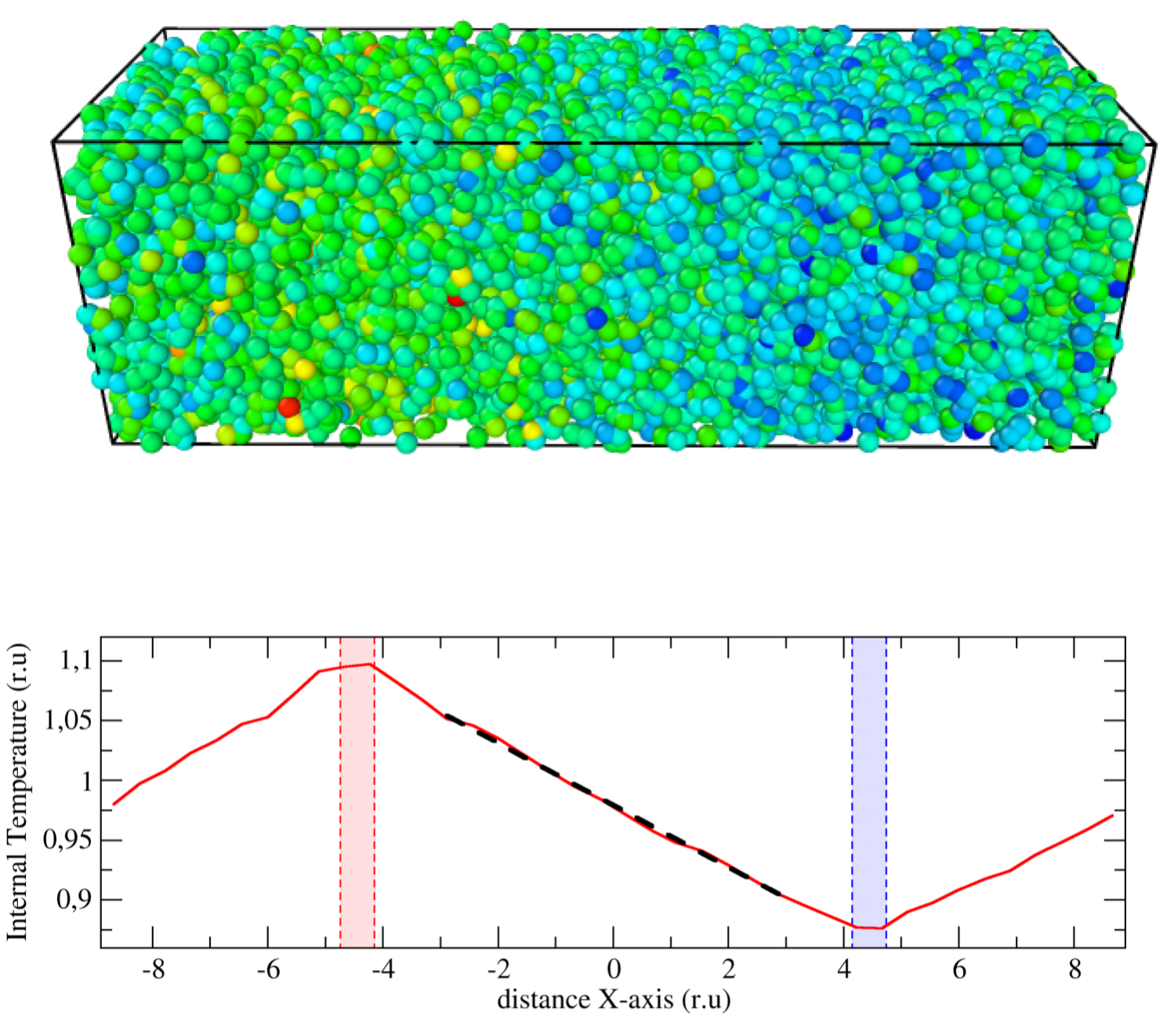 We demonstrate that contrary to general belief, the standard Einstein–Helfand (EH) formulas are valid for the evaluation of transport coefficients of systems containing dissipative and random forces provided that for these mesoscopic systems: (i) the corresponding conservation laws are satisfied, and (ii) the transition probabilities satisfy detailed balance. Dissipative particle dynamics (DPD) and energy-conserving DPD methods (DPDE), for instance, are archetypical of such mesoscopic approaches satisfying these properties. To verify this statement, we have derived a mesoscopic heat flux form for the DPDE method, suitable for the calculation of the thermal conductivity from an EH expression. We have compared EH measurements against non-equilibrium simulation values for different scenarios, including many-body potentials, and have found excellent agreement in all cases. The expressions are valid notably for systems with density- and temperature-dependent potentials, such as the recently developed generalized DPDE method (GenDPDE) [Avalos et al., Phys. Chem. Chem. Phys. 21, 24891, 2019]. We thus demonstrate that traditional EH formulas in equilibrium simulations can be widely used to obtain transport coefficients, provided that the appropriate expression for the associated flux is used.
We demonstrate that contrary to general belief, the standard Einstein–Helfand (EH) formulas are valid for the evaluation of transport coefficients of systems containing dissipative and random forces provided that for these mesoscopic systems: (i) the corresponding conservation laws are satisfied, and (ii) the transition probabilities satisfy detailed balance. Dissipative particle dynamics (DPD) and energy-conserving DPD methods (DPDE), for instance, are archetypical of such mesoscopic approaches satisfying these properties. To verify this statement, we have derived a mesoscopic heat flux form for the DPDE method, suitable for the calculation of the thermal conductivity from an EH expression. We have compared EH measurements against non-equilibrium simulation values for different scenarios, including many-body potentials, and have found excellent agreement in all cases. The expressions are valid notably for systems with density- and temperature-dependent potentials, such as the recently developed generalized DPDE method (GenDPDE) [Avalos et al., Phys. Chem. Chem. Phys. 21, 24891, 2019]. We thus demonstrate that traditional EH formulas in equilibrium simulations can be widely used to obtain transport coefficients, provided that the appropriate expression for the associated flux is used.
- C. Malaspina, M. Lísal, J. P. Larentzos, J. K. Brennan, A. D. Mackie, J. Bonet Avalos, Transport coefficients from Einstein-Helfand relations using standard and energy-conserving dissipative particle dynamics methods, Phys. Chem. Chem. Phys. 25, 12025-12040, 2023. DOI
Adsorption, diffusion, and transport of C1 to C3 alkanes and carbon dioxide in dual-porosity kerogens: Insights from molecular simulations
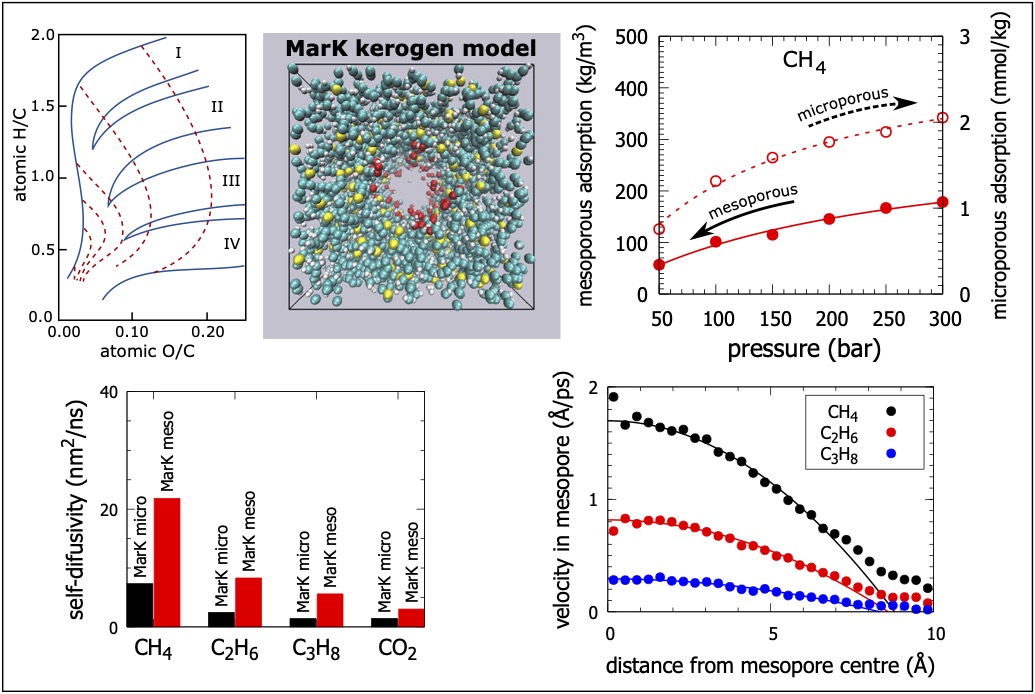 Organic-shale formations are unconventional gas reservoirs with broad pore size distributions. Shale consists of two distinct components: organic matter and clay minerals. The size of pores in the organic matter is mostly concentrated at less than six nanometres and these micropores, and small mesopores provide the majority of adsorption surface area and gas storage volume. In these nanometre-sized pores, the geofluid behaviour becomes significantly different from the bulk behaviour due to the strong solid-fluid interactions and other confinement effects. Understanding the properties of fluids such as methane, ethane, propane and carbon dioxide in narrow shale pores is critical for identifying ways to deploy shale gas technology with reduced environmental impact. Specifically, methane is a proxy of the shale gas and ethane, and propane are minor shale-gas components. Further, carbon dioxide is used for the enhanced shale-gas recovery. We employ molecular-level simulations to explore adsorption, diffusion, and transport of methane, ethane, propane and carbon dioxide in realistic models of organic-shale materials at a representative shale-reservoir temperature and pressures. We first use Hybrid Reverse Monte Carlo with experimental pair distribution functions to build dual-porosity kerogen models corresponding to an immature marine kerogen from the Eagle Ford Play and a mature marine kerogen from the clay-rich Marcellus Play. We then employ Grand Canonical Monte Carlo simulations to study the fluid adsorption in the porous kerogen structures. We complete the adsorption studies by simulating the self-diffusivity, collective diffusivity, and transport diffusivity of the adsorbed fluid molecules in the shale kerogens using equilibrium and non-equilibrium molecular dynamics.
Organic-shale formations are unconventional gas reservoirs with broad pore size distributions. Shale consists of two distinct components: organic matter and clay minerals. The size of pores in the organic matter is mostly concentrated at less than six nanometres and these micropores, and small mesopores provide the majority of adsorption surface area and gas storage volume. In these nanometre-sized pores, the geofluid behaviour becomes significantly different from the bulk behaviour due to the strong solid-fluid interactions and other confinement effects. Understanding the properties of fluids such as methane, ethane, propane and carbon dioxide in narrow shale pores is critical for identifying ways to deploy shale gas technology with reduced environmental impact. Specifically, methane is a proxy of the shale gas and ethane, and propane are minor shale-gas components. Further, carbon dioxide is used for the enhanced shale-gas recovery. We employ molecular-level simulations to explore adsorption, diffusion, and transport of methane, ethane, propane and carbon dioxide in realistic models of organic-shale materials at a representative shale-reservoir temperature and pressures. We first use Hybrid Reverse Monte Carlo with experimental pair distribution functions to build dual-porosity kerogen models corresponding to an immature marine kerogen from the Eagle Ford Play and a mature marine kerogen from the clay-rich Marcellus Play. We then employ Grand Canonical Monte Carlo simulations to study the fluid adsorption in the porous kerogen structures. We complete the adsorption studies by simulating the self-diffusivity, collective diffusivity, and transport diffusivity of the adsorbed fluid molecules in the shale kerogens using equilibrium and non-equilibrium molecular dynamics.
- E. Rezlerová, S. K. Jain, M. Lísal, Adsorption, diffusion, and transport of C1 to C3 alkanes and carbon dioxide in dual-porosity kerogens: Insights from molecular simulations, Energy Fuels 37, 492-508, 2023. DOI