Dynamické odezvy v energetických materiálech
Mikroškálové a mesoškálové jevy (<1 μm) hrají dominantní roli ve výsledných makroskopických vlastnostech široké škály typů materiálů, např. komplexních tekutin či kompozitních materiálů. Prediktivní modelování a simulace odezvy materiálů vyžaduje víceškálové modelování na délkových a časových škálách sahající od atomů až po kontinuum. Modelování a simulace na mesoškále vzhledem k velikosti systému je na jedné straně mimo možnosti molekulárních simulací a na druhé straně simulace na úrovni kontinua nedokáží explicitně zahrnout mikrostrukturu materiálu a spoléhají se tak na fenomenologické modely. Zaměřujeme se na vývoj nových metod pro částicové mesoškálové modelování časo-prostorového uvolňování energie, přenosu tepla, přenosu hmoty a chemické reaktivity energetických materiálů. Naším cílem je vytvořit rigorózní, dobře podložený a obecný výpočetní přístup pro systematické studie zaměřené na identifikaci a charakterizaci základních aspektů dynamické odezvy v energetických materiálech.
Tekutiny v nanoprostoru
Fázové, difuzní a transportní chování tekutin v nanoskopicky omezeném prostoru (mikropóry, mesopóry) nebo na rozhraní povrchů např. grafenu se výrazně liší od chování v objemové fázi. Tato výrazná a často kontraintuivní změna chování tekutin v nanoprostoru je způsobena dominantním efektem interakcí mezi molekulami tekutiny a atomy povrchu. Výsledkem interakcí molekuly tekutiny-atomy povrchu je prostorová nehomogenita hustoty systému, existence specifických fázových přechodů, redukce difuze a transportu např. vodných roztoků na rozhraní hydrofilních povrchů či naopak výrazný nárůst difuze a transportu např. vody v karbonových nanotrubičkách. Pomocí molekulární dynamiky a Monte Carlo simulací se snažíme pochopit, popsat a kvantifikovat vliv nanoprostoru a charakteru interakcí molekuly tekutiny-atomy povrchu na fázové, difuzní a transportní chování tekutin v nanoprostoru. Chování tekutin v nanoprostoru má úzkou vazbu na nanotechnologické obory pracující s porézními materiály či s modifikovanými povrchy.
Povrchové jevy na nanoskopicky modifikovaných površích
Nedávné experimentální studie jasně prokázaly, že modifikace pevných povrchů může mít dramatický vliv na chování adsorbované okolní tekutiny. Ukazuje se, že geometrická či chemická heterogenita povrchů může indukovat nové typy fázových přechodů a kritických jevů, které se kvalitativně liší od povrchových jevů na hladkých podložích. Metodami statistické mechaniky (teorie funkcionálu hustoty, efektivní Hamiltonián, škálování,..) se snažíme vysvětlit fundamentální podstatu těchto jevů a speciálně souvislosti mezi geometrickou charakteristikou daného povrchu, mikroskopickými interakcemi, povrchovými fluktuacemi a měřitelnými vlastnostmi systému (jako je např. adsorpce). Orientujeme se přitom především na systémy, jejichž povrchovou heterogenitu lze charakterizovat na škále odpovídající molekulárním rozměrům. Tento obor je stále ve fázi svého vývoje, ale již prokázal, že pozorované jevy jsou mnohdy kontraintuitivní a nelze je popsat “standardními” postupy. Kromě svého teoretického významu, má tento výzkum i úzkou vazbu na moderní nanotechnologické obory vyvíjející modifikované (např. superhydrofobní) materiály nebo zařízení kontrolující tok extrémně malých objemů kapalin (např. laboratoř na čipu).
Ionic selectivity of NaCl solutions in graphene-based single-digit nanopores
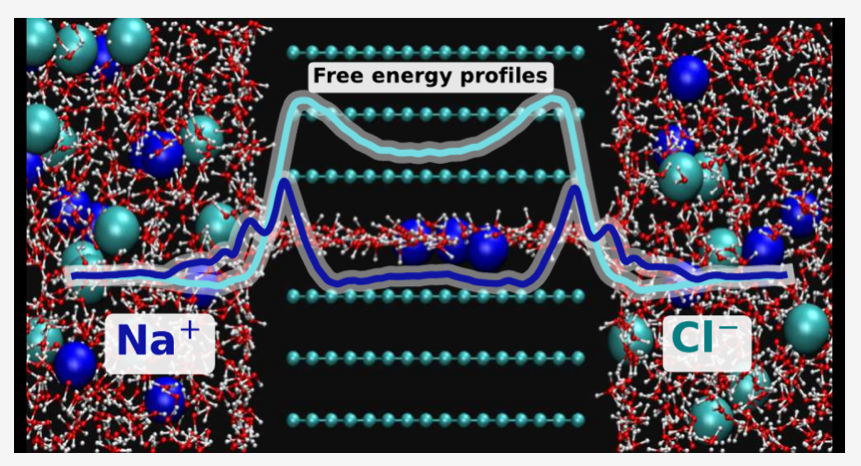 This study provides insights into molecular mechanisms of capacitive deionization (CDI), which has emerged as a promising technique for efficient and sustainable desalination. Through molecular dynamics simulations, we investigate the structure, bonding, and energetics of NaCl aqueous solution in parallel graphene sheets of separation 0.7 or 0.9 nm and lengths 1, 2, or 6 nm. The accelerated weight histogram method implemented in GROMACS was used to compute axial free energy profiles from the reservoir solution to the pore’s center, informing not only about the energetics in the pore’s interior, but also about energy barriers in the transition zones at the pore’s edges, which affect the flow of ions to/through the pores and the charge balance in the pore. We elucidate the roles of surface charge, pore height, and pore length on the ion adsorption, selectivity, and free energy profiles. These findings provide guidance for designing and optimizing CDI systems, thus contributing to the advancement of water treatment technologies and addressing the global water scarcity challenge.
This study provides insights into molecular mechanisms of capacitive deionization (CDI), which has emerged as a promising technique for efficient and sustainable desalination. Through molecular dynamics simulations, we investigate the structure, bonding, and energetics of NaCl aqueous solution in parallel graphene sheets of separation 0.7 or 0.9 nm and lengths 1, 2, or 6 nm. The accelerated weight histogram method implemented in GROMACS was used to compute axial free energy profiles from the reservoir solution to the pore’s center, informing not only about the energetics in the pore’s interior, but also about energy barriers in the transition zones at the pore’s edges, which affect the flow of ions to/through the pores and the charge balance in the pore. We elucidate the roles of surface charge, pore height, and pore length on the ion adsorption, selectivity, and free energy profiles. These findings provide guidance for designing and optimizing CDI systems, thus contributing to the advancement of water treatment technologies and addressing the global water scarcity challenge.
- A. Djeukeng Momo, M. Lísal, M. Předota: Ionic selectivity of NaCl solutions in graphene-based single-digit nanopores. Langmuir 42, 15392-15406, 2026. DOI
Multiple Meniscus Depinning Transitions in Open Capillary Slits
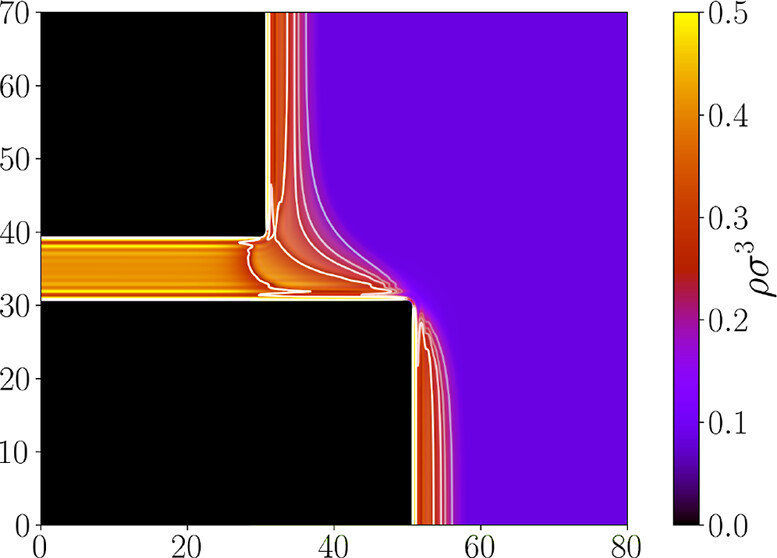 We investigate meniscus depinning transitions in confined fluids within semi-infinite capillary slits with asymmetric open edges using both macroscopic theory and classical density functional theory (DFT). We identify four distinct condensed morphologies depending on the pinning state of the meniscus and determine the conditions for transitions between them as functions of the slit geometry and contact angle. Our analysis shows that all depinning transitions are continuous, being second order for partial wetting and third order for complete wetting or drying. Microscopic DFT calculations confirm the predicted phase behavior and reveal important packing and finite-size effects, especially under complete wetting conditions where adsorption layers effectively reduce the accessible slit width. The results provide a systematic description of pinning-depinning phenomena in confined fluids and their dependence on confinement geometry and wetting properties.
We investigate meniscus depinning transitions in confined fluids within semi-infinite capillary slits with asymmetric open edges using both macroscopic theory and classical density functional theory (DFT). We identify four distinct condensed morphologies depending on the pinning state of the meniscus and determine the conditions for transitions between them as functions of the slit geometry and contact angle. Our analysis shows that all depinning transitions are continuous, being second order for partial wetting and third order for complete wetting or drying. Microscopic DFT calculations confirm the predicted phase behavior and reveal important packing and finite-size effects, especially under complete wetting conditions where adsorption layers effectively reduce the accessible slit width. The results provide a systematic description of pinning-depinning phenomena in confined fluids and their dependence on confinement geometry and wetting properties.
- J. Janek, A.O. Parry, A. Malijevský: Multiple Meniscus Depinning Transitions in Open Capillary Slits. J. Phys. Chem. B 129(51), 13325–13338, 2026. DOI
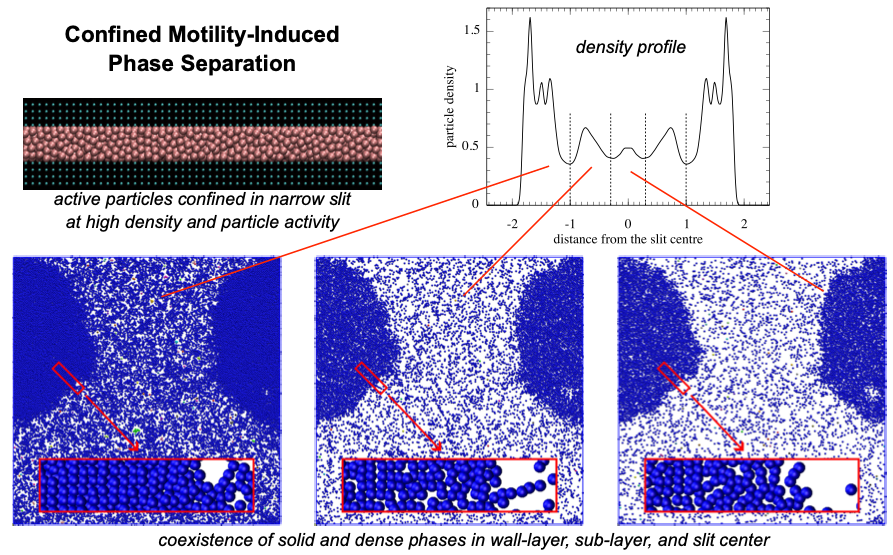
Motility-induced phase separations in confined active particle systems
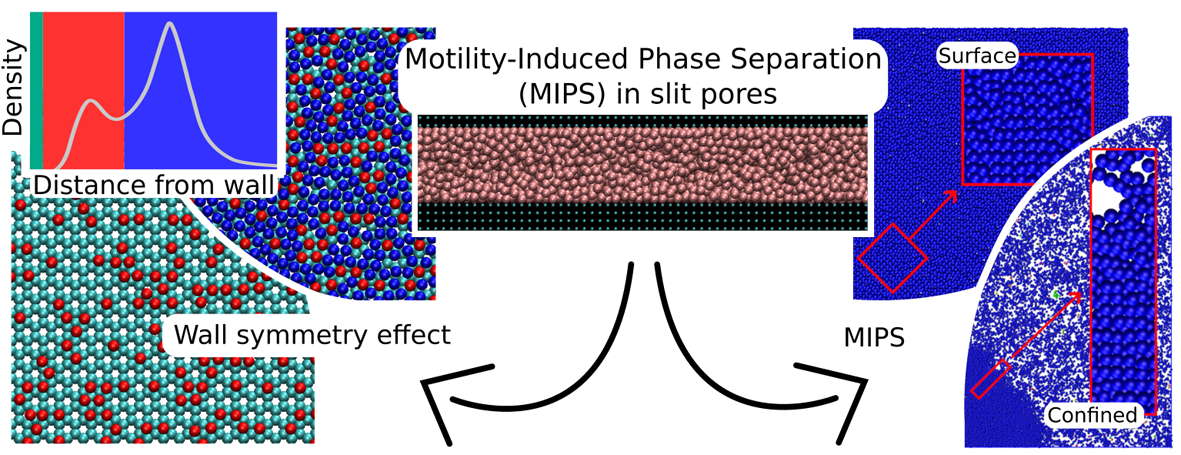 Active Brownian particles (ABPs) serve as a versatile model for synthetic active matter, such as self-propelled colloids, combining persistent propulsion with Brownian motion. When confined, ABPs exhibit propulsion-induced wall accumulation, a phenomenon that can be exploited in microfluidics and lab-on-a-chip applications. In addition, at high confined densities and particle activities, ABPs display distinct structural phenomena, including confined and surface motility-induced phase separations (MIPSs). Confined MIPS manifests as the coexistence of solid-like and dense phases, whereas surface MIPS involves the emergence of high-density clustering structures near the confining walls. The formation of these solid-like and high-density clustering-structure states, along with their structures, is further influenced by wall roughness. We investigate ABPs in slit pores using overdamped Langevin dynamics simulations at a liquid particle density and a particle activity exceeding the bulk MIPS threshold. We examine their wall accumulation and MIPS behavior as functions of slit width and wall roughness. Furthermore, we contrast these confined ABPs with their equilibrium counterparts, confined fluid particles capable of completely wetting the slit walls.
Active Brownian particles (ABPs) serve as a versatile model for synthetic active matter, such as self-propelled colloids, combining persistent propulsion with Brownian motion. When confined, ABPs exhibit propulsion-induced wall accumulation, a phenomenon that can be exploited in microfluidics and lab-on-a-chip applications. In addition, at high confined densities and particle activities, ABPs display distinct structural phenomena, including confined and surface motility-induced phase separations (MIPSs). Confined MIPS manifests as the coexistence of solid-like and dense phases, whereas surface MIPS involves the emergence of high-density clustering structures near the confining walls. The formation of these solid-like and high-density clustering-structure states, along with their structures, is further influenced by wall roughness. We investigate ABPs in slit pores using overdamped Langevin dynamics simulations at a liquid particle density and a particle activity exceeding the bulk MIPS threshold. We examine their wall accumulation and MIPS behavior as functions of slit width and wall roughness. Furthermore, we contrast these confined ABPs with their equilibrium counterparts, confined fluid particles capable of completely wetting the slit walls.
- K. Šindelka, K. Aim, M. Lísal: Motility-induced phase separations in confined active particle systems. Fluid Ph. Equilib. 605, 114690, 2026. DOI
Ion pairing restructures the electric double layer in water-in-salt electrolytes on graphite and alters capacitance
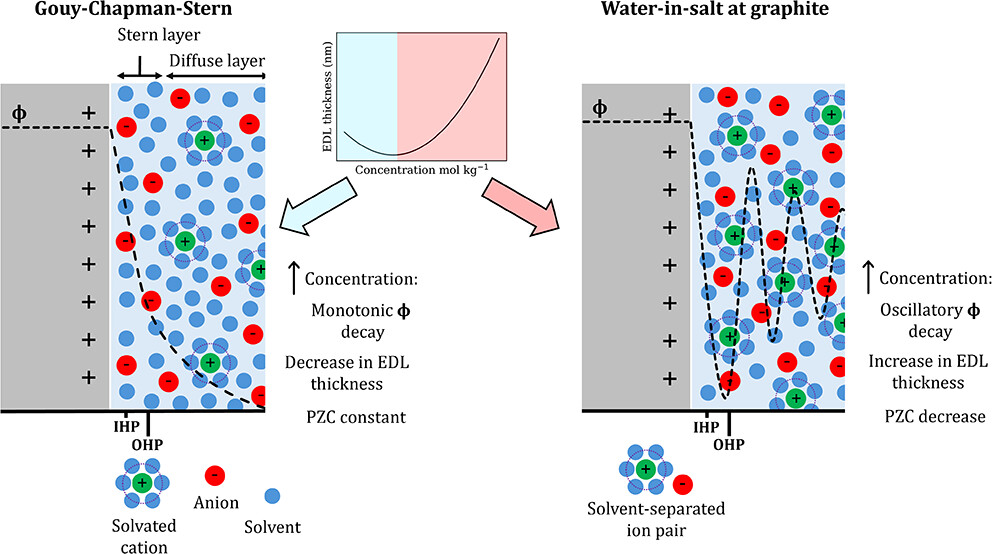 The structure and thickness of the electrical double layer (EDL) at carbon electrodes strongly influence electrochemical performance, yet remain poorly understood in concentrated aqueous electrolytes. Here we combine classical and quantum-mechanical molecular dynamics simulations with experimental analyses to resolve the interfacial organization of aqueous LiCl from dilute to water-in-salt (WiS) (1−20 mol/kg) concentrations at graphitic electrodes, and compare with electrochemical differential-capacitance measurements from which the potential of zero charge (PZC) is obtained. We uncover a concentration-driven restructuring of the EDL: below 6 mol/kg, solvated Li+ dominates the outer Helmholtz plane (OHP), but at higher concentrations, co-adsorption of Cl− through solvent-separated ion pairs enforces a near 1:1 Li/Cl ratio at the interface. This transition expands the effective EDL thickness, redistributes the interfacial potential drop, and drives a decrease in the PZC, matching the trend inferred from differential-capacitance measurements on electrolyte-graphite interfaces. Capacitance calculations reveal that while both EDL and quantum contributions vary strongly with concentration, their opposing trends make the total capacitance appear nearly constant for a pristine graphite slab comprising four atomic layers; for other electrodes where quantum capacitance is not limiting and electrode-internal series contributions are small, the total capacitance would more directly reflect the concentration dependence of the EDL capacitance. Solvent-separated ion pairing is identified as the key driver of non-monotonic EDL behavior in LiCl WiS electrolytes, establishing design considerations for tuning interfacial capacitance and stability in next-generation carbon-based aqueous energy storage systems.
The structure and thickness of the electrical double layer (EDL) at carbon electrodes strongly influence electrochemical performance, yet remain poorly understood in concentrated aqueous electrolytes. Here we combine classical and quantum-mechanical molecular dynamics simulations with experimental analyses to resolve the interfacial organization of aqueous LiCl from dilute to water-in-salt (WiS) (1−20 mol/kg) concentrations at graphitic electrodes, and compare with electrochemical differential-capacitance measurements from which the potential of zero charge (PZC) is obtained. We uncover a concentration-driven restructuring of the EDL: below 6 mol/kg, solvated Li+ dominates the outer Helmholtz plane (OHP), but at higher concentrations, co-adsorption of Cl− through solvent-separated ion pairs enforces a near 1:1 Li/Cl ratio at the interface. This transition expands the effective EDL thickness, redistributes the interfacial potential drop, and drives a decrease in the PZC, matching the trend inferred from differential-capacitance measurements on electrolyte-graphite interfaces. Capacitance calculations reveal that while both EDL and quantum contributions vary strongly with concentration, their opposing trends make the total capacitance appear nearly constant for a pristine graphite slab comprising four atomic layers; for other electrodes where quantum capacitance is not limiting and electrode-internal series contributions are small, the total capacitance would more directly reflect the concentration dependence of the EDL capacitance. Solvent-separated ion pairing is identified as the key driver of non-monotonic EDL behavior in LiCl WiS electrolytes, establishing design considerations for tuning interfacial capacitance and stability in next-generation carbon-based aqueous energy storage systems.
- H.O. Wood, F. Zhou, J. Dočkal, M. Lísal, F. Moučka, S. Kaewmorakot, R.A.W. Dryfe, P. Carbone: Ion pairing restructures the electric double layer in water-in-salt electrolytes on graphite and alters capacitance. J. Phys. Chem. C 130, 5965-5978, 2026. DOI
Using dissipative particle dynamics to model polymeric systems
 This chapter reviews the principles and applications of dissipative particle dynamics (DPD) as a coarse‑grained simulation technique for polymeric systems. It begins by outlining the multiscale structural and dynamical complexity of polymers, which spans many orders of magnitude in time and length and makes fully atomistic simulations impractical. DPD is introduced as a mesoscale, momentum‑conserving method in which soft beads represent groups of atoms or Kuhn segments and interact via conservative, dissipative, and random forces chosen to preserve hydrodynamics and impose a thermostat. The Groot-Warren formulation is described in detail, including soft repulsive bead-bead interactions, spring and FENE bond potentials for flexible and semiflexible chains, and additional segment-segment repulsions to suppress unphysical chain crossing in entangled melts. Electrostatic interactions in ion‑containing polymers are then incorporated through smeared charge distributions and Ewald‑type treatments, with discussion of charge assignment strategies for quenched and annealed polyelectrolytes and the delicate balance between Coulombic and excluded‑volume forces. Parametrization strategies are presented that map DPD repulsion parameters onto Flory-Huggins interaction parameters and experimental compressibilities, with extensions to mixtures of beads of different sizes and densities. The second part of the chapter surveys DPD applications: polymer solutions and melts across concentration regimes; self‑assembly of block copolymers into micelles, vesicles, and complex nanoparticles; electrostatic co‑assembly of oppositely charged polyelectrolytes into interpolyelectrolyte complexes; and polymer behavior at interfaces and under 1D-3D confinement, including adsorption, slit pores, and emulsion droplets. The authors close by highlighting both the power and limitations of DPD, emphasizing the need for physically sound coarse‑graining and parametrization, and pointing to future developments such as improved electrostatics, adaptive‑resolution schemes, and data‑driven parameter optimization to extend DPD’s role in designing advanced polymeric materials.
This chapter reviews the principles and applications of dissipative particle dynamics (DPD) as a coarse‑grained simulation technique for polymeric systems. It begins by outlining the multiscale structural and dynamical complexity of polymers, which spans many orders of magnitude in time and length and makes fully atomistic simulations impractical. DPD is introduced as a mesoscale, momentum‑conserving method in which soft beads represent groups of atoms or Kuhn segments and interact via conservative, dissipative, and random forces chosen to preserve hydrodynamics and impose a thermostat. The Groot-Warren formulation is described in detail, including soft repulsive bead-bead interactions, spring and FENE bond potentials for flexible and semiflexible chains, and additional segment-segment repulsions to suppress unphysical chain crossing in entangled melts. Electrostatic interactions in ion‑containing polymers are then incorporated through smeared charge distributions and Ewald‑type treatments, with discussion of charge assignment strategies for quenched and annealed polyelectrolytes and the delicate balance between Coulombic and excluded‑volume forces. Parametrization strategies are presented that map DPD repulsion parameters onto Flory-Huggins interaction parameters and experimental compressibilities, with extensions to mixtures of beads of different sizes and densities. The second part of the chapter surveys DPD applications: polymer solutions and melts across concentration regimes; self‑assembly of block copolymers into micelles, vesicles, and complex nanoparticles; electrostatic co‑assembly of oppositely charged polyelectrolytes into interpolyelectrolyte complexes; and polymer behavior at interfaces and under 1D-3D confinement, including adsorption, slit pores, and emulsion droplets. The authors close by highlighting both the power and limitations of DPD, emphasizing the need for physically sound coarse‑graining and parametrization, and pointing to future developments such as improved electrostatics, adaptive‑resolution schemes, and data‑driven parameter optimization to extend DPD’s role in designing advanced polymeric materials.
- K. Procházka, K. Šindelka, Z. Limpouchová, M. Lísal: Using dissipative particle dynamics to model polymeric systems. Computational Methods for the Multiscale Modeling of Soft Matter, Part One - Soft matter modeling methods, pp. 3-35, edited by P. Carbone and N. Clarke; Methods in Molecular and Materials Modeling Book Series, series editor R. Catlow; Elsevier, 2026. ISBN: 978-0-443-27314-8. DOI
Complete Wetting and Drying at Sinusoidal Walls
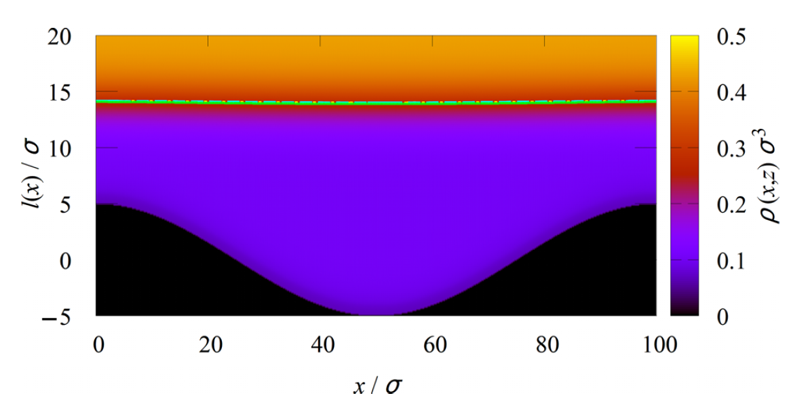 We investigate complete wetting and drying at sinusoidally corrugated walls, focusing on how wall geometry and the range of intermolecular interactions affect the growth and shape of adsorbed fluid layers near saturation. We consider two limiting cases: complete drying at purely repulsive walls with short-ranged interactions and complete wetting at attractive walls with long-ranged van der Waals forces. Using nonlocal interfacial Hamiltonian theory and a sharp-kink approximation, we derive scaling relations for the mean interface height and interface corrugation as functions of the wall amplitude and wavelength. We show that short-ranged and long-ranged systems display qualitatively different scaling regimes and asymptotic behavior. The theoretical predictions are tested using fully microscopic density functional theory calculations, which confirm the predicted scaling laws with excellent quantitative agreement, including for strongly corrugated substrates. The results provide a detailed description of wetting and drying phenomena at structured surfaces and demonstrate the important role played by both geometry and the nature of microscopic interactions.
We investigate complete wetting and drying at sinusoidally corrugated walls, focusing on how wall geometry and the range of intermolecular interactions affect the growth and shape of adsorbed fluid layers near saturation. We consider two limiting cases: complete drying at purely repulsive walls with short-ranged interactions and complete wetting at attractive walls with long-ranged van der Waals forces. Using nonlocal interfacial Hamiltonian theory and a sharp-kink approximation, we derive scaling relations for the mean interface height and interface corrugation as functions of the wall amplitude and wavelength. We show that short-ranged and long-ranged systems display qualitatively different scaling regimes and asymptotic behavior. The theoretical predictions are tested using fully microscopic density functional theory calculations, which confirm the predicted scaling laws with excellent quantitative agreement, including for strongly corrugated substrates. The results provide a detailed description of wetting and drying phenomena at structured surfaces and demonstrate the important role played by both geometry and the nature of microscopic interactions.
- A. Malijevský, M. Pospíšil, M. Magociová, J. Janek: Complete Wetting and Drying at Sinusoidal Walls. Phys. Rev. E 112, 015502, 2025. DOI
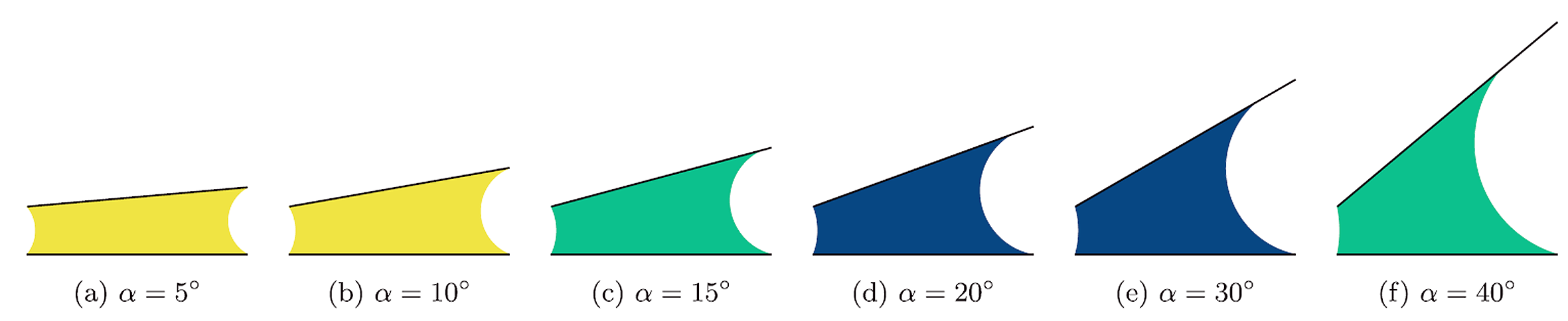
Three Types of Condensation in Open Wedges
We investigate capillary condensation in open wedges formed by two nonparallel walls of finite length connected to a bulk gas reservoir. In contrast to infinitely long wedges, where filling occurs continuously, condensation in open wedges is always first-order and exhibits remarkably rich phase behavior controlled by the wedge opening angle, aspect ratio, and Young contact angle. Using a macroscopic thermodynamic description based on meniscus geometry and free-energy minimization, we identify three distinct condensation states: single-pinning (SP), semi-double-pinning (SDP), and double-pinning (DP). These states differ in the configuration and pinning of the menisci separating the condensed liquid from the surrounding gas. We derive Kelvin-like equations governing each condensation regime, determine the conditions for transitions between them, and construct global phase diagrams revealing reentrant behavior and continuous higher-order depinning transitions between condensation states. The analysis further provides asymptotic scaling laws in several important limits, including narrow wedges, very long systems, and the approach to the condensation threshold.
- J. Janek, A. Malijevský: Three Types of Condensation in Open Wedges. Phys. Rev. E 111, 045507, 2025. DOI
Capillary Condensation between Parallel Walls of Unequal Length
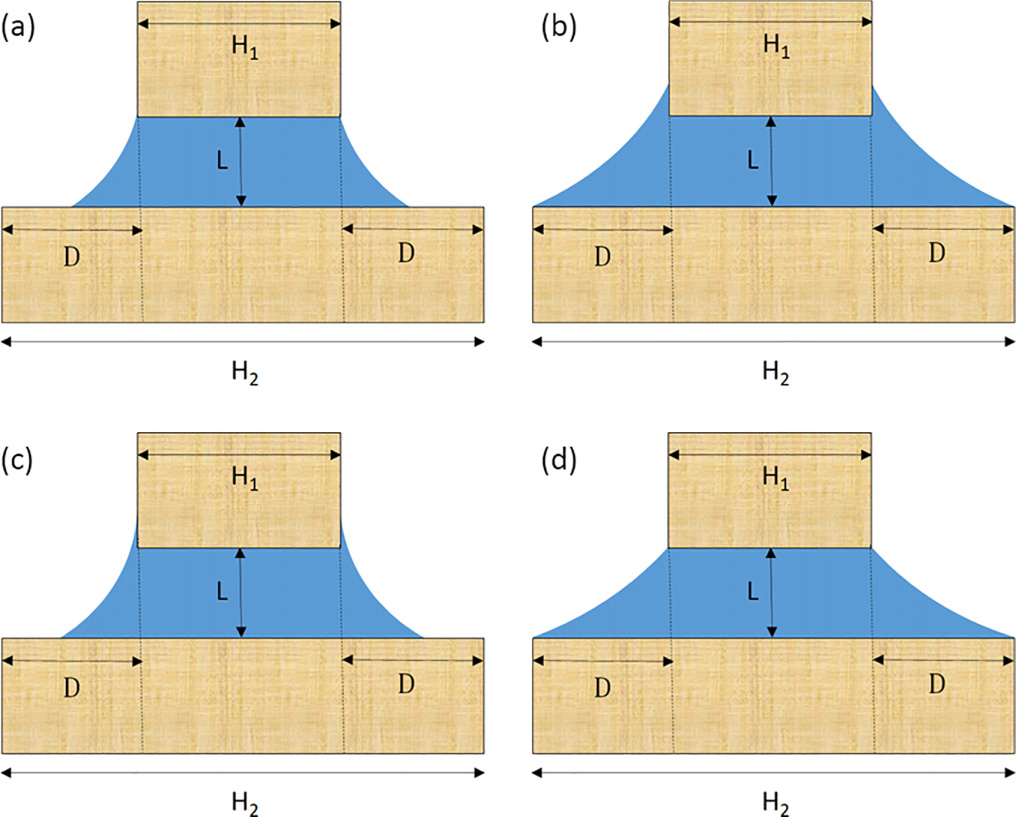 This work develops a macroscopic theory of capillary condensation in asymmetric slit geometries formed by two parallel walls of unequal lengths. Unlike the classical infinite slit, where condensation is described by a single Kelvin equation, finite and truncated walls give rise to multiple competing condensation morphologies controlled by wall geometry and wettability. Using geometric arguments and free-energy minimization based on the concept of edge contact angles, four distinct condensation states are identified: the 1+ state, where menisci are pinned at the edges of the shorter wall; the 1− state, pinned at the longer wall; the 2 state, where both walls pin the menisci; and the 0 state, where the menisci meet the walls at equilibrium contact angles without edge pinning. For each morphology, Kelvin-like equations are derived that determine the undersaturation at which condensation occurs. The resulting phase behavior is governed by three dimensionless parameters: the reduced upper-wall length H1/L, the relative overhang D/L, and the Young contact angle θ. The analysis reveals two fundamental organizing principles: a geometric separatrix at D = L that distinguishes 0-condensation from 2-condensation, and the wedge-filling threshold θ = π/4, which separates a rich four-state regime from a simpler two-state regime. Global phase diagrams show how increasing contact angle progressively suppresses the 0 and 1− condensation states, enlarges the no-condensation region, and eventually leaves only the 1+ and 2 states for θ > π/4. Analytical asymptotic expressions are obtained for all phase boundaries and condensation limits, including saturation conditions and scaling laws near geometric transitions. The theory demonstrates how finite-size effects and asymmetric confinement qualitatively modify capillary phase behavior relative to classical slit models.
This work develops a macroscopic theory of capillary condensation in asymmetric slit geometries formed by two parallel walls of unequal lengths. Unlike the classical infinite slit, where condensation is described by a single Kelvin equation, finite and truncated walls give rise to multiple competing condensation morphologies controlled by wall geometry and wettability. Using geometric arguments and free-energy minimization based on the concept of edge contact angles, four distinct condensation states are identified: the 1+ state, where menisci are pinned at the edges of the shorter wall; the 1− state, pinned at the longer wall; the 2 state, where both walls pin the menisci; and the 0 state, where the menisci meet the walls at equilibrium contact angles without edge pinning. For each morphology, Kelvin-like equations are derived that determine the undersaturation at which condensation occurs. The resulting phase behavior is governed by three dimensionless parameters: the reduced upper-wall length H1/L, the relative overhang D/L, and the Young contact angle θ. The analysis reveals two fundamental organizing principles: a geometric separatrix at D = L that distinguishes 0-condensation from 2-condensation, and the wedge-filling threshold θ = π/4, which separates a rich four-state regime from a simpler two-state regime. Global phase diagrams show how increasing contact angle progressively suppresses the 0 and 1− condensation states, enlarges the no-condensation region, and eventually leaves only the 1+ and 2 states for θ > π/4. Analytical asymptotic expressions are obtained for all phase boundaries and condensation limits, including saturation conditions and scaling laws near geometric transitions. The theory demonstrates how finite-size effects and asymmetric confinement qualitatively modify capillary phase behavior relative to classical slit models.
- A. Malijevský: Capillary Condensation between Parallel Walls of Unequal Length. Phys. Rev. E 112, 065502, 2025. DOI
Interpretable machine-learning enhanced parametrization methodology for Pluronics–water mixtures in DPD simulations
 Dissipative particle dynamics (DPD) is an incredibly powerful tool for simulating the behavior of structured fluids. However, identifying the appropriate model parameters to accurately replicate physical properties remains a challenge. This study showcases the benefits of integrating machine learning techniques into the top-down parameterization of Pluronic systems. The proposed workflow outlines a data-driven approach to accurately determine model parameters tailored to various Pluronic systems. Gaussian process regression (GPR)-based surrogate models effectively replicate the results of DPD simulations, delivering faster responses that streamline parameter optimization and enable the calibration of Pluronic systems against experimental data. Although DPD simulations provide valuable insight, their high computational cost, due to extensive simulations and post-processing, presents a challenge. The GPR-based surrogate model addresses this by modeling the relationships between input parameters and output properties. SHAP (SHapley additive exPlanations) analysis enhances model interpretability, providing deeper insights into the relationships and causal mechanisms between the input parameters and the predicted properties. The combination of GPR and SHAP analysis provides an interpretable machine learning approach, enabling a more efficient optimization process and reducing the need for exhaustive simulations. This work lays a foundation for generalizing the parameterization process across Pluronic systems and conditions, such as varying temperatures, by incorporating additional DPD model input parameters.
Dissipative particle dynamics (DPD) is an incredibly powerful tool for simulating the behavior of structured fluids. However, identifying the appropriate model parameters to accurately replicate physical properties remains a challenge. This study showcases the benefits of integrating machine learning techniques into the top-down parameterization of Pluronic systems. The proposed workflow outlines a data-driven approach to accurately determine model parameters tailored to various Pluronic systems. Gaussian process regression (GPR)-based surrogate models effectively replicate the results of DPD simulations, delivering faster responses that streamline parameter optimization and enable the calibration of Pluronic systems against experimental data. Although DPD simulations provide valuable insight, their high computational cost, due to extensive simulations and post-processing, presents a challenge. The GPR-based surrogate model addresses this by modeling the relationships between input parameters and output properties. SHAP (SHapley additive exPlanations) analysis enhances model interpretability, providing deeper insights into the relationships and causal mechanisms between the input parameters and the predicted properties. The combination of GPR and SHAP analysis provides an interpretable machine learning approach, enabling a more efficient optimization process and reducing the need for exhaustive simulations. This work lays a foundation for generalizing the parameterization process across Pluronic systems and conditions, such as varying temperatures, by incorporating additional DPD model input parameters.
- N. Lauriello, D.N. Ponnana, Z. Ma, K. Šindelka, A. Buffo, G. Boccardo, D. Marchisio, W. Pan: Interpretable machine-learning enhanced parametrization methodology for Pluronics–water mixtures in DPD simulations, Soft Matter 21, 5833-5851, 2025. DOI
Dissipative particle dynamics as a computational tool to detect the morphology-rheology interplay in Pluronic F68/water mixtures: A promising drug carrier
Pluronics, also known as poloxamers, are amphiphilic triblock copolymers widely employed in drug delivery systems due to their tunable self-assembly and biocompatibility. Among them, Pluronic F68 (Poloxamer 188) exhibits thermoresponsive behavior in aqueous solution, forming ordered supramolecular structures at high concentrations and temperatures. In this work, we investigate the morphological and rheological properties of a 45 wt% Pluronic F68 aqueous system at different temperatures through a combination of experimental and computational approaches. Rheological measurements and Small-Angle X-ray Scattering (SAXS) confirm the formation of a body-centered cubic (BCC) structure at higher temperatures and highlight the emergence of viscoelastic solid-like behavior. To support and extend these findings, Dissipative Particle Dynamics (DPD) simulations are employed to model the nanostructure evolution and the impact of temperature on self-assembly and material properties. This integrated approach provides a consistent framework to characterize the temperature-induced transition from fluid-like to solid-like states and sets the groundwork for future simulation studies incorporating drug cargo. The results offer valuable insights into the design of thermoresponsive drug delivery systems and demonstrate the potential of DPD in capturing complex structure–property relationships in amphiphilic polymer systems.

- N. Lauriello, N. A. Di Spirito, K. Šindelka, G. Boccardo, R. Pasquino, N. Grizzuti, D. Marchisio: Dissipative particle dynamics as a computational tool to detect the morphology-rheology interplay in Pluronic F68/water mixtures: A promising drug carrier. J. Colloid Interface Sci. 700, 138525, 2025. DOI
Confined active particles: Wall accumulation and correspondence between active and fluid systems
Active Brownian particles (ABPs) are a generic model for synthetic active matter systems, such as active colloids, characterized by their ability to self-propel while also exhibiting Brownian motion. Under confinement, ABPs display propulsion-induced wall accumulation, a behavior that can be harnessed for microfluidic and lab-on-a-chip applications. We investigate ABPs confined in slit pores using overdamped Langevin dynamics and study their wall accumulation as a function of particle activity and slit width, at both gas and liquid densities. Furthermore, we examine the correspondence between confined ABPs and their equilibrium counterparts, confined fluid particles capable of completely wetting the slit walls.
- K. Šindelka, A. Gadermeteva, M. Lísal: Confined active particles: Wall accumulation and correspondence between active and fluid systems. Soft Matter 21, 7544-7564, 2025. DOI
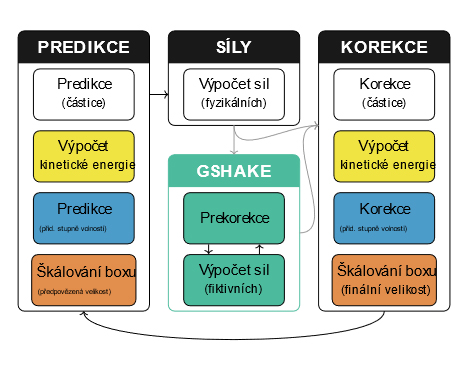
Od prediktorů a SHAKE metody k jednotnému integračnímu schématu pro molekulární dynamiku
Molekulární dynamika (MD) poskytuje detailní vhled do dějů na molekulární úrovni. V průběhu let jejího vývoje bylo vyvinuto mnoho postupů integrace pohybových rovnic, které tvoří její základ. Postupem času se vytříbilo několik algoritmů, které jsou považovány za nejlepší, a počáteční variabilita integračních metod tak vzala za své. Přestože jsou používané metody mnohdy opravdu adekvátní, občas se vyskytnou i úlohy, kde by mohlo být výhodné použít alternativní přístup. Cílem této práce tak bylo ukázat alternativní univerzální přístup k formulaci integračních algoritmů pro MD. Navržené „jednotné integrační schéma“ pro MD založené na prediktorech obsahuje značnou část variability, která se časem vytratila.
Prvním krokem k jednotnému schématu byla adaptace tradiční SHAKE metody pro integraci molekulárních systémů s pevnými vazbami do integračního schématu prediktor–korektor, dalším krokem pak zahrnutí metody pro předpověď rychlostí (time-reversible velocity predictor). V rámci tohoto přístupu je pak možné použít jak tradiční Verletovu integrační metodu, tak nedávno vylepšené metody Gearovy. Výsledné jednotné integrační schéma bylo testováno na dvou jednoduchých modelech a dvou realistických MD systémech: na SPC/E modelu vody a na modelu jednoduché iontové kapaliny (1-ethyl-3-methylimidazolium tetrafluorborátu). Pomocí navrženého algoritmu byly vygenerovány trajektorie v mikrokanonickém, kanonickém (Nosé–Hoover) a isobarickém (MTK) souboru. Když se v novém schématu použila jako základ Verletova metoda, byly získané výsledky ve velmi přesvědčivé shodě s výsledky získanými konvenčními algoritmy. Ukázalo se také, že v některých případech může být výhodné použít Gearovy metody, které jsou vyššího řádu. Naše univerzální schéma nabízí ve srovnání se standardními přístupy podstatně jednoduší způsob, jak navrhovat nové integrační algoritmy, což může zjednodušit údržbu simulačních programů pro MD. Výsledné algoritmy se navíc skvěle hodí i pro masivně paralelní implementaci.
- J. Janek: From predictors and SHAKE toward unified integration scheme for molecular dynamics. J. Chem. Phys. 162, 084103, 2025. DOI
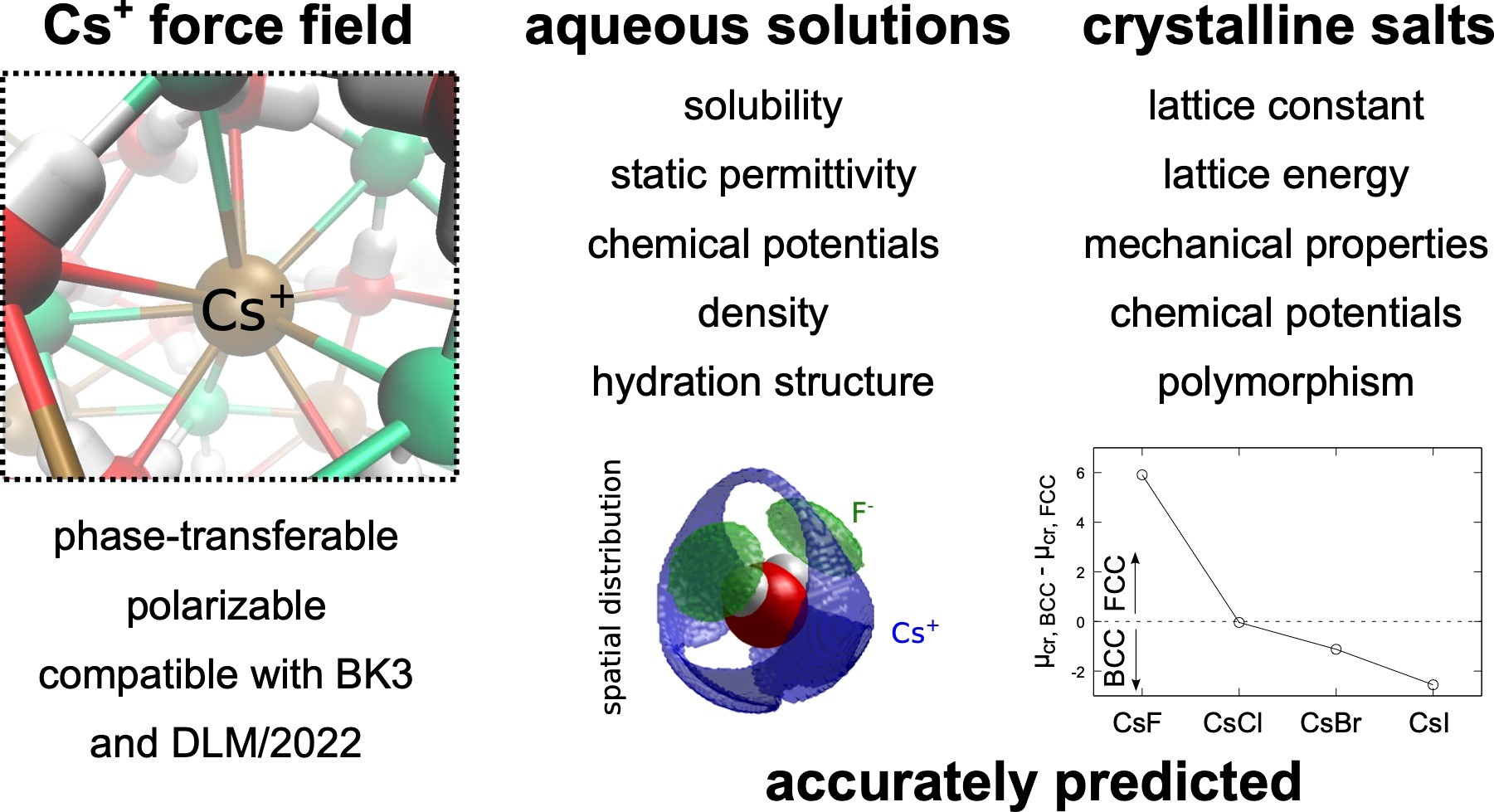
Molecular simulations of cesium halide aqueous solutions and crystalline salts using phase-transferable polarizable force fields
Cesium, an important alkali metal halide, exhibits distinct behavior necessitating accurate modeling of its interactions. We present a refined polarizable force field for Cs+ ions, extending the set of DLM/2022-BK3 FFs, to improve predictions of cesium halide properties. The refined model accurately captures structural properties, phase transitions, and concentration-dependent properties of aqueous solutions of CsF, CsCl, CsBr, and CsI, including high concentrations. Structural insights reveal variations in hydration numbers and the formation of contact ion pairs, especially pronounced with larger counterions. Notably, the force fields accurately predict aqueous solubilities, highlighting their accuracy in both ion-water and ion-ion interactions. While primarily designed for single salt solutions, these models offer broad applicability, extending to complex mixtures. Our findings underscore the reliability and versatility of the developed Cs+ model and the DLM/2022-BK3 force fields, which provides a valuable tool for studying alkali metal halide systems.
- J. Dočkal, P. Mimrová, M. Lísal, F. Moučka: Molecular simulations of cesium halide aqueous solutions and crystalline salts using phase-transferable polarizable force fields. J. Mol. Liq. 424, 127121, 2025. DOI
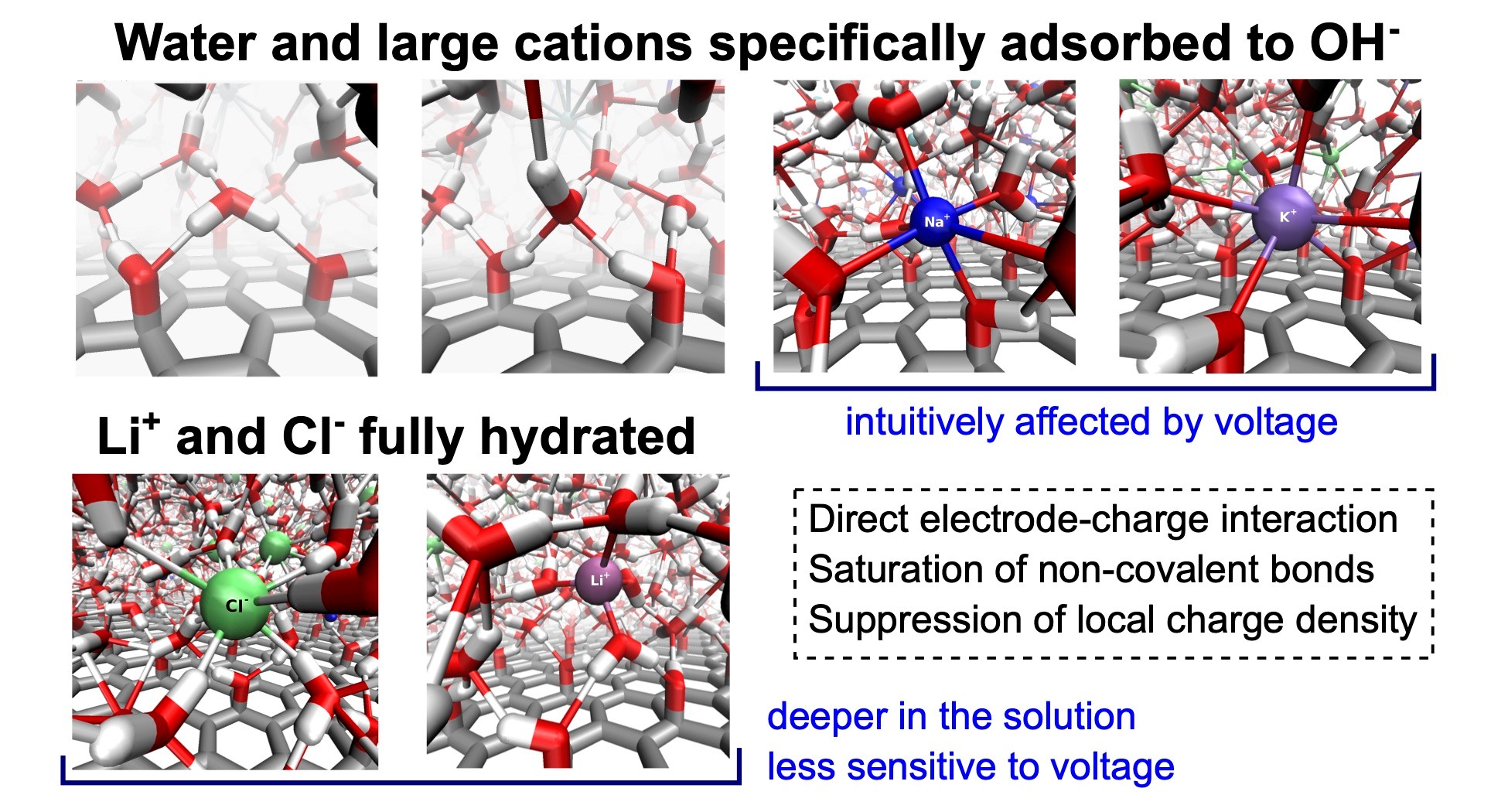
Understanding interfacial structure and preferential adsorption in mixed alkali-halide electrolytes at graphene oxide electrodes by constant potential molecular dynamics simulations
Graphene oxide (GO) is a promising material that finds use in electrochemical applications. Therefore, understanding the microscopic behavior of electrolytes in contact with GO electrodes is important. In this work, we focus on a detailed description and explanation of the structure, adsorption behavior, and self-diffusion of aqueous solutions of single-salt and mixed-cation alkali metal chlorides in the vicinity of hydroxylated GO electrodes under normal thermodynamic conditions and varying interelectrode voltages. We performed molecular dynamics simulations of the solutions constrained between planar GO electrodes using the constant potential method. We analyzed several structural properties, including profiles of atomic density, charge density, characteristics of the network of non-covalent bonds, and in-plane and transverse self-diffusion, all as functions of the distance from the GO surface. We discuss and explain the behavior of all these properties in detail as a result of three driving forces: (i) the direct electrode-solution interactions, (ii) the tendency of the solutions to saturate the network of non-covalent bonds, and (iii) the tendency of the system to suppress local charge accumulation in any region larger than typical interparticle distances. The existence of hydroxyl groups here greatly enhances the direct electrode-solution interactions, causing qualitative differences in the structure, including a different arrangement of the adsorption layers of ions in comparison to the solutions at graphene electrodes.
- J. Dočkal, E. Rezlerová, M. Předota, M. Lísal, F. Moučka: Understanding interfacial structure and preferential adsorption in mixed alkali-halide electrolytes at graphene oxide electrodes by constant potential molecular dynamics simulations. J. Mol. Liq. 424, 127078, 2025. DOI
Modeling temperature-dependent transport properties in dissipative particle dynamics: A top-down coarse-graining toward realistic dynamics at the mesoscale
Dissipative particle dynamics (DPD) is a widespread computational tool to simulate the behavior of soft matter and liquids in and out of equilibrium. Although there are many applications in which the effect of temperature is relevant, most of the DPD studies have been carried out at a fixed system temperature. Therefore, this work investigates how to incorporate the effect of system temperature variation within the DPD model to capture realistic temperature-dependent system properties. In particular, this work focuses on the relationship between temperature and transport properties, and therefore, an extended DPD model for transport properties prediction is employed. Transport properties, unlike the equilibrium properties, are often overlooked despite their significant influence on the flow dynamics of non-isothermal mesoscopic systems. Moreover, before simulating the response of the system induced by a temperature change, it is important to first estimate transport properties at a certain temperature. Thus here, the same fluid is simulated across different temperature conditions using isothermal DPD with the aim to identify a temperature-dependent parametrization methodology, capable of ensuring the correctness of both equilibrium and dynamical properties. Liquid water is used as a model system for these analyses. This work proposes a temperature-dependent form of the extended DPD model where both conservative and non-conservative interaction parameters incorporate the variation of the temperature. The predictions provided by our simulations are in excellent agreement with experimental data.
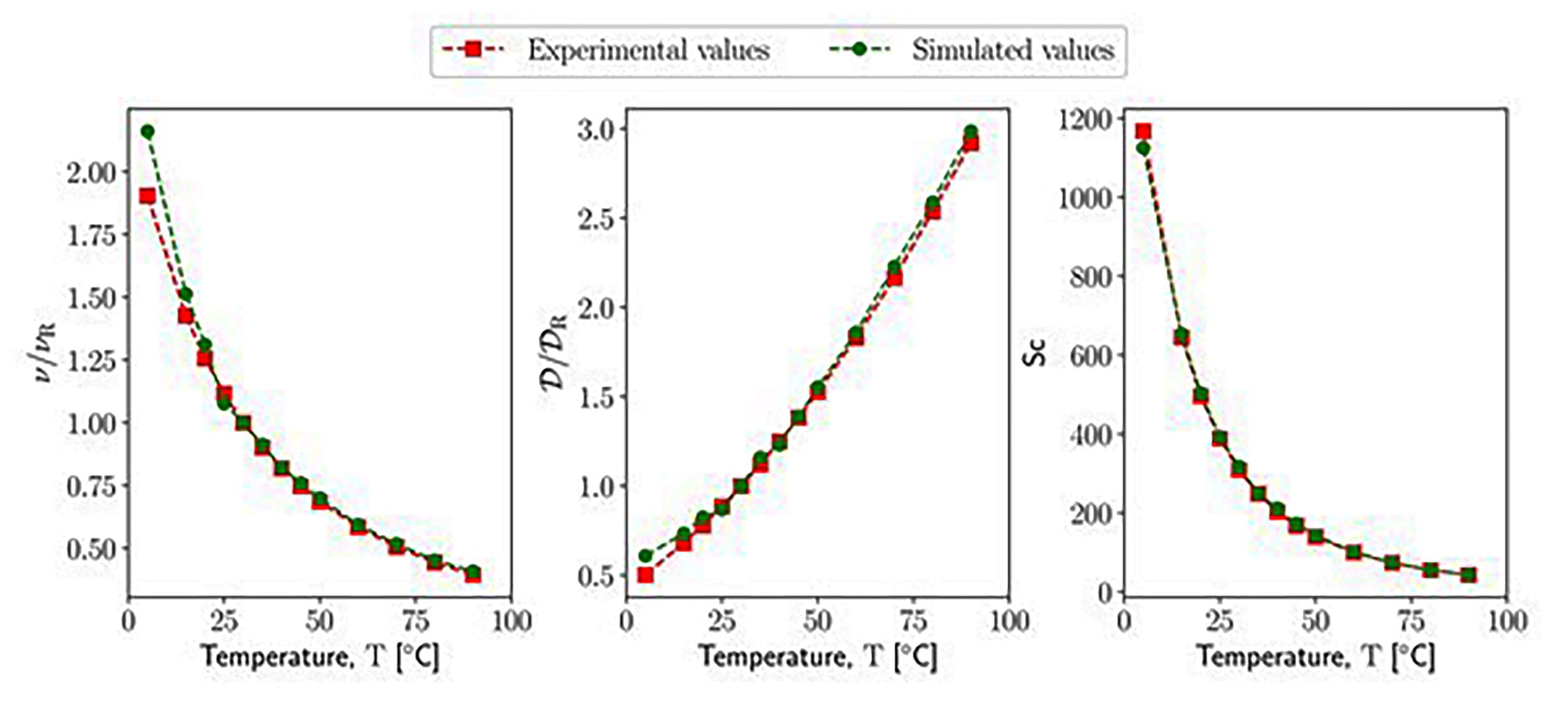
Comparison of the transport properties of water at different temperatures between experimental data and the results of DPD simulations
- N. Lauriello, M. Lísal, G. Boccardo, D. Marchisio, A. Buffo: Modeling temperature-dependent transport properties in dissipative particle dynamics: A top-down coarse-graining toward realistic dynamics at the mesoscale. J. Chem. Phys. 161(3), 034112, 2024. DOI
Generalized energy-conserving dissipative particle dynamics with mass transfer: Coupling between energy and mass exchange
The complete description of energy and material transport within the Generalized energy-conserving dissipative particle dynamics with mass transfer (GenDPDE-M) methodology is presented. In particular, the dynamic coupling between mass and energy is incorporated into the GenDPDE-M, which was previously introduced with dynamically decoupled fluxes (J. Bonet Avalos et al., J. Chem. Theory Comput. 18(12), 7639–7652, 2022). From a theoretical perspective, we have derived the appropriate Fluctuation-Dissipation theorems along with Onsager’s reciprocal relations, suitable for mesoscale models featuring this coupling. Equilibrium and non-equilibrium simulations are performed to demonstrate the internal thermodynamic consistency of the method, as well as the ability to capture the Ludwig–Soret effect and tune its strength through the mesoscopic parameters. In view of the completeness of the presented approach, GenDPDE-M is the most general Lagrangian method to deal with complex fluids and systems at the mesoscale, where thermal agitation is relevant.
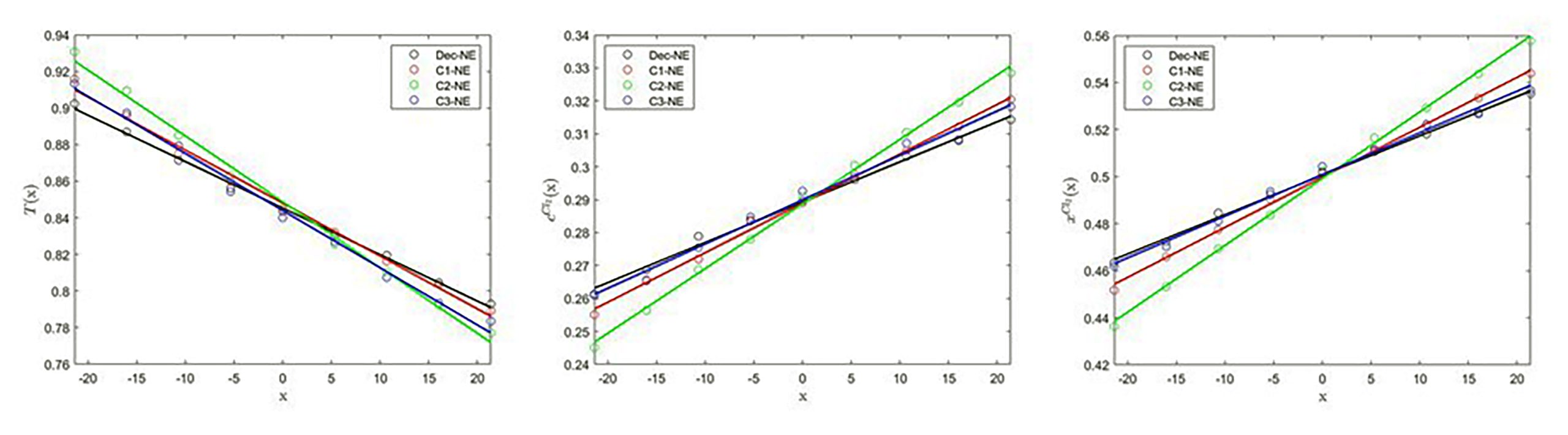 The x-profiles of the particle temperature (left), and the number concentration (middle) and molar fraction (right) of Cl2 for the coarse-grain equimolar Ar/Cl2 binary mixture
The x-profiles of the particle temperature (left), and the number concentration (middle) and molar fraction (right) of Cl2 for the coarse-grain equimolar Ar/Cl2 binary mixture
- G. Colella, A.D. Mackie, J.P. Larentzos, J.K. Brennan, M. Lísal, J. Bonet Avalos: Generalized energy-conserving dissipative particle dynamics with mass transfer: Coupling between energy and mass exchange. J. Non-Equilib. Thermodyn. 49, 347-375, 2024. DOI
Kelvin equation for bridging transitions
see Research Achievements of the Institute
- A. Malijevský, M. Pospíšil: Kelvin equation for bridging transitions. Phys. Rev. E 109(3 March), 034801, 2024. DOI
Capillary condensation between nonparallel walls
see Research Achievements of the Institute.
- A. Malijevský, J. Janek: Capillary condensation between nonparallel walls. Phys. Rev. E 109, 054801, 2024. DOI
Structure and self-diffusivity of mixed-cation electrolytes between neutral and charged graphene sheets
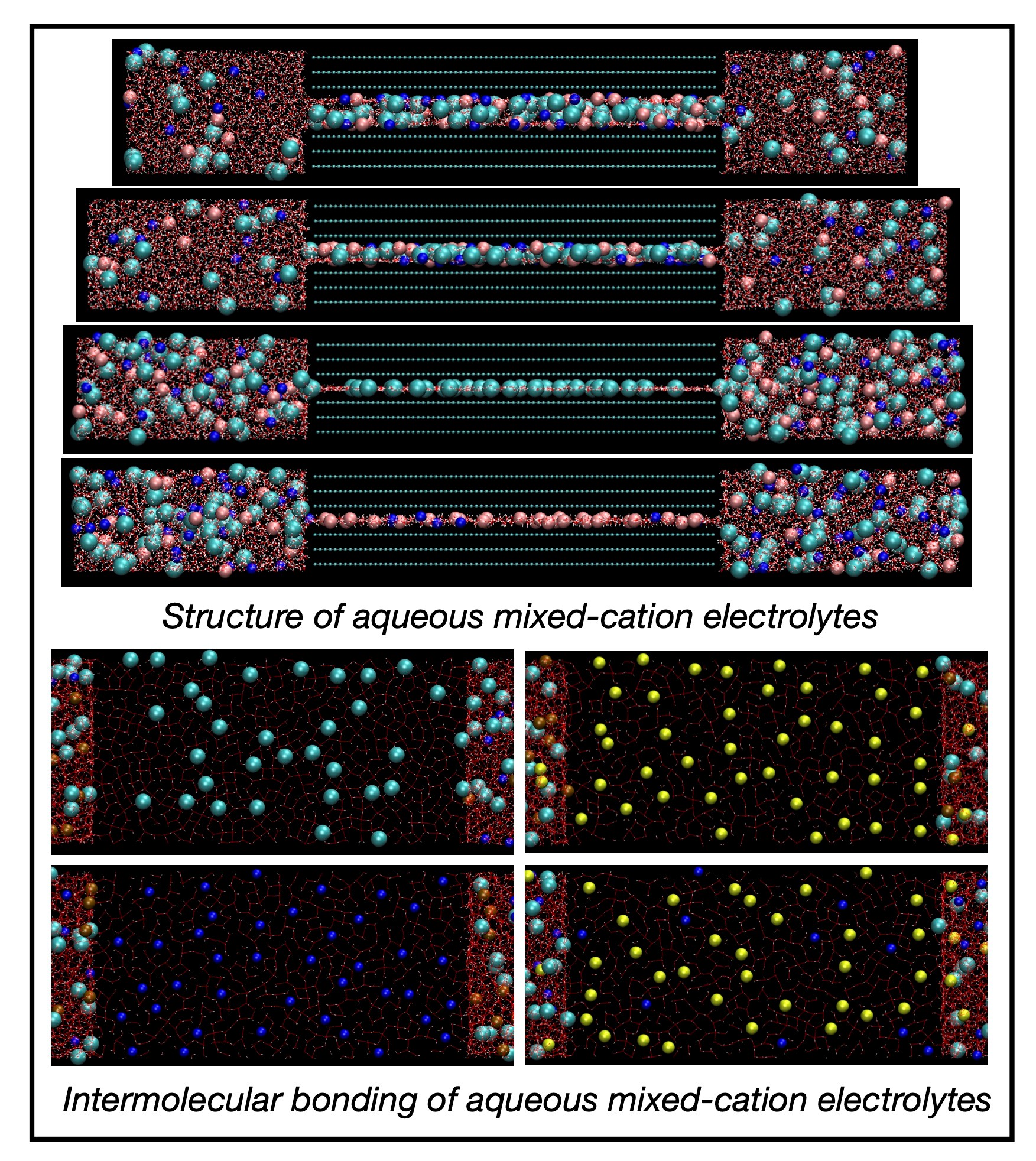 Graphene-based applications, such as supercapacitors or capacitive deionization, take place in an aqueous environment, and they benefit from molecular-level insights into the behavior of aqueous electrolyte solutions in single-digit graphene nanopores with a size comparable to a few molecular diameters. Under single-digit graphene nanoconfinement (smallest dimension < 2 nm), water and ions behave drastically different than in the bulk. Most aqueous electrolytes in graphene-based applications as well as in nature contain a mix of electrolytes. We study several prototypical aqueous mixed alkali-chloride electrolytes containing an equimolar fraction of Li/Na, Li/K, or Na/K cations confined between neutral and positively or negatively charged parallel graphene sheets. The strong hydration shell of small Li+ vs a larger Na+ or large K+ with weaker or weak hydration shells affects the interplay between the ions’ propensity to hydrate or dehydrate under the graphene nanoconfinement and the strength of the ion–graphene interactions mediated by confinement-induced layered water. We perform molecular dynamics simulations of the confined mixed-cation electrolytes using the effectively polarizable force field for electrolyte–graphene systems and focus on a relation between the electrochemical adsorption and structural properties of the water molecules and ions and their diffusion behavior. The simulations show that the one-layer nanoslits have the biggest impact on the ions’ adsorption and the water and ions’ diffusion. The positively charged one-layer nanoslits only allow for Cl− adsorption and strengthen the intermolecular bonding, which along with the ultrathin confinement substantially reduces the water and Cl− diffusion. In contrast, the negatively charged one-layer nanoslits only allow for the adsorption of weakly hydrated Na+ or K+ and substantially break up the non-covalent bond network, which leads to the enhancement of the water and Na+ or K+ diffusion up to or even above the bulk diffusion. In wider nanoslits, cations adsorb closer to the graphene surfaces than Cl−’s with preferential adsorption of a weakly hydrated cation over a strongly hydrated cation. The positive graphene charge has an intuitive effect on the adsorption of weakly hydrated Na+’s or K+’s and Cl−’s and a counterintuitive effect on the adsorption of strongly hydrated Li+’s. On the other hand, the negative surface charge has an intuitive effect on the adsorption of both types of cations and only mild intuitive or counterintuitive effects on the Cl− adsorption. The diffusion of water molecules and ions confined in the wider nanoslits is reduced with respect to the bulk diffusion, more for the positive graphene charge, which strengthened the intermolecular bonding, and less for the negative surface charge, which weakened the non-covalent bond network.
Graphene-based applications, such as supercapacitors or capacitive deionization, take place in an aqueous environment, and they benefit from molecular-level insights into the behavior of aqueous electrolyte solutions in single-digit graphene nanopores with a size comparable to a few molecular diameters. Under single-digit graphene nanoconfinement (smallest dimension < 2 nm), water and ions behave drastically different than in the bulk. Most aqueous electrolytes in graphene-based applications as well as in nature contain a mix of electrolytes. We study several prototypical aqueous mixed alkali-chloride electrolytes containing an equimolar fraction of Li/Na, Li/K, or Na/K cations confined between neutral and positively or negatively charged parallel graphene sheets. The strong hydration shell of small Li+ vs a larger Na+ or large K+ with weaker or weak hydration shells affects the interplay between the ions’ propensity to hydrate or dehydrate under the graphene nanoconfinement and the strength of the ion–graphene interactions mediated by confinement-induced layered water. We perform molecular dynamics simulations of the confined mixed-cation electrolytes using the effectively polarizable force field for electrolyte–graphene systems and focus on a relation between the electrochemical adsorption and structural properties of the water molecules and ions and their diffusion behavior. The simulations show that the one-layer nanoslits have the biggest impact on the ions’ adsorption and the water and ions’ diffusion. The positively charged one-layer nanoslits only allow for Cl− adsorption and strengthen the intermolecular bonding, which along with the ultrathin confinement substantially reduces the water and Cl− diffusion. In contrast, the negatively charged one-layer nanoslits only allow for the adsorption of weakly hydrated Na+ or K+ and substantially break up the non-covalent bond network, which leads to the enhancement of the water and Na+ or K+ diffusion up to or even above the bulk diffusion. In wider nanoslits, cations adsorb closer to the graphene surfaces than Cl−’s with preferential adsorption of a weakly hydrated cation over a strongly hydrated cation. The positive graphene charge has an intuitive effect on the adsorption of weakly hydrated Na+’s or K+’s and Cl−’s and a counterintuitive effect on the adsorption of strongly hydrated Li+’s. On the other hand, the negative surface charge has an intuitive effect on the adsorption of both types of cations and only mild intuitive or counterintuitive effects on the Cl− adsorption. The diffusion of water molecules and ions confined in the wider nanoslits is reduced with respect to the bulk diffusion, more for the positive graphene charge, which strengthened the intermolecular bonding, and less for the negative surface charge, which weakened the non-covalent bond network.
- E. Rezlerová, F. Moučka, M. Předota, M. Lísal: Structure and self-diffusivity of mixed-cation electrolytes between neutral and charged graphene sheets, J. Chem. Phys. 160, 094701, 2024. DOI
Green-Kubo expressions for transport coefficients from dissipative particle dynamics simulations revisited
 This article addresses the debate about the correct application of Green–Kubo expressions for transport coefficients from dissipative particle dynamics simulations. We demonstrate that the Green–Kubo expressions are valid provided that (i) the dynamic model conserves the physical property, whose transport is studied, and (ii) the fluctuations satisfy detailed balance. As a result, the traditional expressions used in molecular dynamics can also be applied to dissipative particle dynamics simulations. However, taking the calculation of the shear viscosity as a paradigmatic example, a random contribution, whose strength scales as 1/dt1/2, with dt the timestep, can cause difficulties if the stress tensor is not separated into the different contributions. We compare our expression to that of Ernst and Brito (M. H. Ernst and R. Brito, Europhys. Lett. 73, 183-189, 2006), which arises from a diametrically different perspective. We demonstrate that the two expressions are completely equivalent and find exactly the same result both analytically and numerically. We show that the differences are not due to the lack of time-reversibility but instead from a preaveraging of the random contributions. Despite the overall validity of Green–Kubo expressions, we find that the Einstein–Helfand relations (D. C. Malaspina et al. Phys. Chem. Chem. Phys. 25, 12025-12040, 2023) do not suffer from the need to decompose the stress tensor and can readily be used with a high degree of accuracy. Consequently, Einstein–Helfand relations should be seen as the preferred method to calculate transport coefficients from dissipative particle dynamics simulations.
This article addresses the debate about the correct application of Green–Kubo expressions for transport coefficients from dissipative particle dynamics simulations. We demonstrate that the Green–Kubo expressions are valid provided that (i) the dynamic model conserves the physical property, whose transport is studied, and (ii) the fluctuations satisfy detailed balance. As a result, the traditional expressions used in molecular dynamics can also be applied to dissipative particle dynamics simulations. However, taking the calculation of the shear viscosity as a paradigmatic example, a random contribution, whose strength scales as 1/dt1/2, with dt the timestep, can cause difficulties if the stress tensor is not separated into the different contributions. We compare our expression to that of Ernst and Brito (M. H. Ernst and R. Brito, Europhys. Lett. 73, 183-189, 2006), which arises from a diametrically different perspective. We demonstrate that the two expressions are completely equivalent and find exactly the same result both analytically and numerically. We show that the differences are not due to the lack of time-reversibility but instead from a preaveraging of the random contributions. Despite the overall validity of Green–Kubo expressions, we find that the Einstein–Helfand relations (D. C. Malaspina et al. Phys. Chem. Chem. Phys. 25, 12025-12040, 2023) do not suffer from the need to decompose the stress tensor and can readily be used with a high degree of accuracy. Consequently, Einstein–Helfand relations should be seen as the preferred method to calculate transport coefficients from dissipative particle dynamics simulations.
- D. C. Malaspina, M. Lísal, J. P. Larentzos, J. K. Brennan, A. D. Mackie, J. Bonet Avalos, Green-Kubo expressions for transport coefficients from dissipative particle dynamics simulations revisited. Phys. Chem. Chem. Phys. 26, 1328-1339, 2024. DOI
Structure of aqueous alkali metal halide electrolyte solutions from molecular simulations of phase-transferable polarizable models
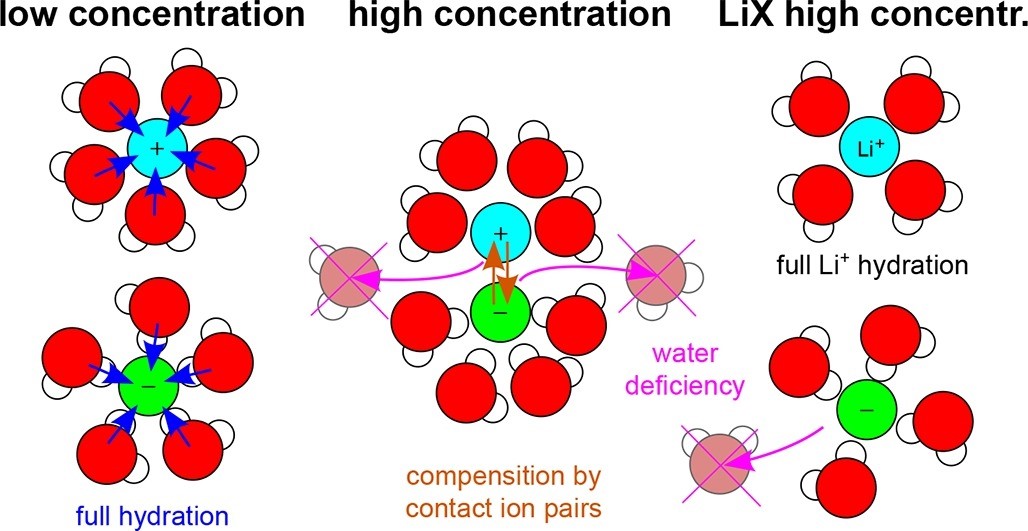 The structure of aqueous solutions of alkali metal halides is studied under ambient thermodynamic conditions and concentrations from infinite dilution to supersaturation using molecular dynamics simulations of phase-transferable polarizable models. The results of the solution densities, radial distribution functions, 3D spatial distribution functions, the properties of hydrogen and other noncovalent bonds in solutions, hydration numbers, coordination numbers, numbers of contact cation-anion pairs, and other statistics of the number of ions hydrated simultaneously by a shared water molecule are systematically presented. In particular, the results show and quantify how the strengths of the hydration bonds of different ions vary and how the hydration numbers decrease with increasing concentration in parallel with an increase in the number of contact cation-anion pairs. In most cases, they completely compensate for the loss of water-ion bonds by an increase in cation-anion bonds. The exceptions are solutions based on the Li+ cation, which retain a solid hydration shell even at high concentrations. This behavior is conceptualized based on three imaginary driving forces: the first dominating at low concentrations and causing full hydration of the ions, the second representing a lack of water necessary for full hydration of the ions and increasing with increasing concentration, and the third attracting counterions to the water-unoccupied sites of the hydration shells and also increasing with concentration. This concept can be used not only to understand the structural behavior of homogeneous electrolytes in thermodynamic equilibrium but also to study phenomena that involve preferential adsorption of ions on electrodes, nanochannels, or porous materials. The data obtained for the number and strength of hydration bonds and ion pairs can also be used in further studies to elucidate the diffusion behavior, viscosity, and conductivity of aqueous electrolyte solutions.
The structure of aqueous solutions of alkali metal halides is studied under ambient thermodynamic conditions and concentrations from infinite dilution to supersaturation using molecular dynamics simulations of phase-transferable polarizable models. The results of the solution densities, radial distribution functions, 3D spatial distribution functions, the properties of hydrogen and other noncovalent bonds in solutions, hydration numbers, coordination numbers, numbers of contact cation-anion pairs, and other statistics of the number of ions hydrated simultaneously by a shared water molecule are systematically presented. In particular, the results show and quantify how the strengths of the hydration bonds of different ions vary and how the hydration numbers decrease with increasing concentration in parallel with an increase in the number of contact cation-anion pairs. In most cases, they completely compensate for the loss of water-ion bonds by an increase in cation-anion bonds. The exceptions are solutions based on the Li+ cation, which retain a solid hydration shell even at high concentrations. This behavior is conceptualized based on three imaginary driving forces: the first dominating at low concentrations and causing full hydration of the ions, the second representing a lack of water necessary for full hydration of the ions and increasing with increasing concentration, and the third attracting counterions to the water-unoccupied sites of the hydration shells and also increasing with concentration. This concept can be used not only to understand the structural behavior of homogeneous electrolytes in thermodynamic equilibrium but also to study phenomena that involve preferential adsorption of ions on electrodes, nanochannels, or porous materials. The data obtained for the number and strength of hydration bonds and ion pairs can also be used in further studies to elucidate the diffusion behavior, viscosity, and conductivity of aqueous electrolyte solutions.
- J. Dočkal, P. Mimrová, M. Lísal, F. Moučka, Structure of aqueous alkali metal halide electrolyte solutions from molecular simulations of phase-transferable polarizable models. J. Mol. Liq. 394, 123797, 2024. DOI
Structure and self-diffusivity of alkali-halide electrolytes in neutral and charged graphene nanochannels
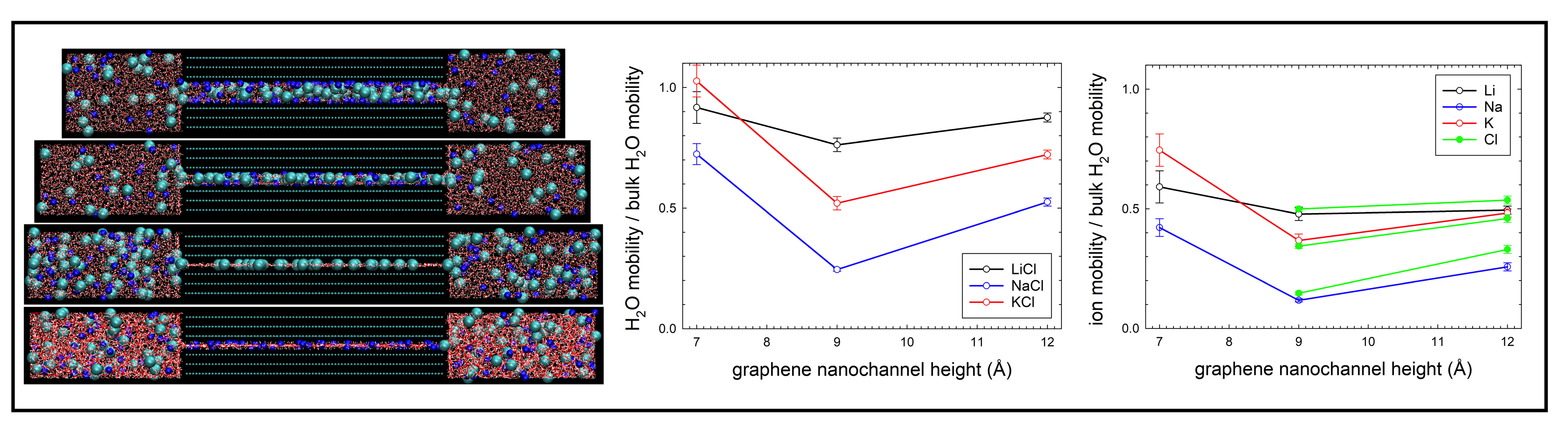 Understanding the microscopic behavior of aqueous electrolyte solutions in graphene-based ultrathin nanochannels is important in nanofluidic applications such as water purification, fuel cells, and molecular sensing. Under extreme confinement (< 2 nm), the properties of water and ions differ drastically from those in the bulk phase. We studied the structural and diffusion behavior of prototypical aqueous solutions of electrolytes (LiCl, NaCl, and KCl) confined in both neutral and positively-, and negatively-charged graphene nanochannels. We performed molecular dynamics simulations of the solutions in the nanochannels with either one, two- or three-layer water structures using the effectively polarisable force field for graphene.
Understanding the microscopic behavior of aqueous electrolyte solutions in graphene-based ultrathin nanochannels is important in nanofluidic applications such as water purification, fuel cells, and molecular sensing. Under extreme confinement (< 2 nm), the properties of water and ions differ drastically from those in the bulk phase. We studied the structural and diffusion behavior of prototypical aqueous solutions of electrolytes (LiCl, NaCl, and KCl) confined in both neutral and positively-, and negatively-charged graphene nanochannels. We performed molecular dynamics simulations of the solutions in the nanochannels with either one, two- or three-layer water structures using the effectively polarisable force field for graphene.
We analyzed the structure and intermolecular bond network of the confined solutions along with their relation to the self-diffusivity of water and ions. The simulations show that Na and K cations can more easily rearrange their solvation shells under the graphene nanoconfinement and adsorb on the graphene surfaces or dissolve in the confinement-induced layered water than the Li cation. The negative surface charge together with the presence of ions orient water molecules with hydrogens towards the graphene surfaces, which in turn weakens the intermolecular bond network. The one-layer nanochannels have the biggest effect on the water structure and intermolecular bonding as well as on the adsorption of ions with only co-ions entering these nanochannels. The self-diffusivity of confined water is strongly reduced with respect to the bulk water and decreases with diminishing nanochannel heights except for the negatively-charged one-layer nanochannel. The self-diffusivity of ions also decreases with the reducing the nanochannel heights except for the self-diffusivity of cations in the negatively-charged one-layer nanochannel, evidencing cooperative diffusion of confined water and ions. Due to the significant break-up of the intermolecular bond network in the negatively-charged one-layer nanochannel, self-diffusion coefficients of water and cations exceed those for the two- and three-layer nanochannels and become comparable to the bulk values.
- E. Rezlerová, F. Moučka, M. Předota, M. Lísal, Structure and self-diffusivity of alkali-halide electrolytes in neutral and charged graphene nanochannels, Phys. Chem. Chem. Phys. 25, 21579-21594, 2023. DOI
Development of an automated reliable method to compute transport properties from DPD equilibrium simulations: Application to simple fluids
D issipative particle dynamics (DPD) is a promising candidate technique for modeling the rheological properties of soft matter systems. However, several methodological issues inhibit its exploitation as a computational rheology tool. In this work, we focus on the development of an automated method to compute transport properties from equilibrium simulation with particular attention to the assessment of the Green-Kubo approach reliability and computational feasibility for a large set of simple DPD systems with increasing Schmidt number. Furthermore, we investigate the time step size dependency of dynamic properties and the role of different time integration schemes. In particular, we assess the performance of the Shardlow-splitting algorithm against the most popular modified velocity-Verlet algorithm. We consider, for the first time, the application of the Shardlow-splitting algorithm to the transverse DPD thermostat in different friction regimes relying on systematic numerical experiments. In addition, we make use of these findings to perform a multi-parametric study aiming to investigate the Schmidt number relationship with the effective friction coefficient.
issipative particle dynamics (DPD) is a promising candidate technique for modeling the rheological properties of soft matter systems. However, several methodological issues inhibit its exploitation as a computational rheology tool. In this work, we focus on the development of an automated method to compute transport properties from equilibrium simulation with particular attention to the assessment of the Green-Kubo approach reliability and computational feasibility for a large set of simple DPD systems with increasing Schmidt number. Furthermore, we investigate the time step size dependency of dynamic properties and the role of different time integration schemes. In particular, we assess the performance of the Shardlow-splitting algorithm against the most popular modified velocity-Verlet algorithm. We consider, for the first time, the application of the Shardlow-splitting algorithm to the transverse DPD thermostat in different friction regimes relying on systematic numerical experiments. In addition, we make use of these findings to perform a multi-parametric study aiming to investigate the Schmidt number relationship with the effective friction coefficient.
- N. Lauriello, G. Boccardo, D. Marchisio, M. Lísal, A. Buffo, Development of an automated reliable method to compute transport properties from DPD equilibrium simulations: Application to simple fluids, Comput. Phys. Commun. 291, 108843, 2023. DOI
Interakce monovrstev kationtové povrchově aktivní látky a mastného alkoholu s přirozeným povrchem lidských vlasů: Postřehy z dynamiky disipativních částic
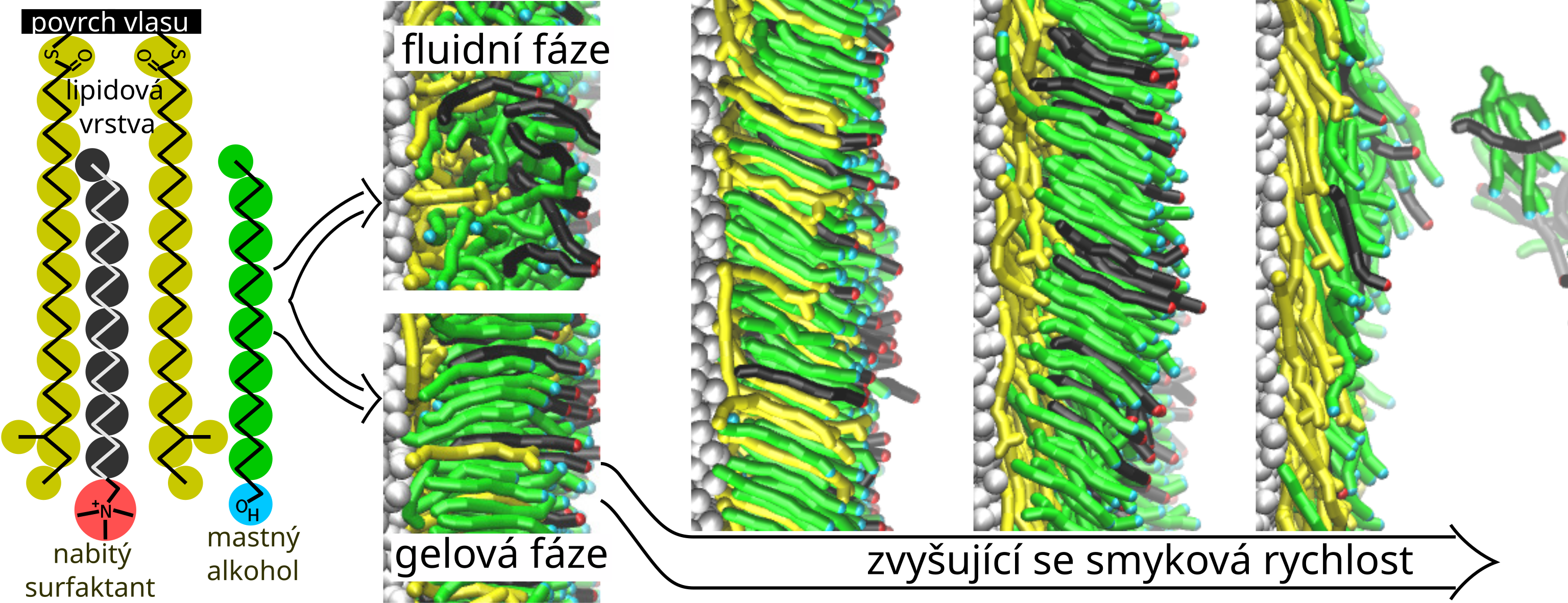 Lamely tvořené mastnými alkoholy a nabitými surfaktanty jsou běžnou součástí tekutých kosmetických přípravků. Pomocí mesoskopického modelování jsme studovali, jak monovrstvy pocházející z odpovídajících dvouvrstev v tekutých kosmetických přípravcích interagují s povrchem lidského vlasu. Prováděli jsme rovnovážné i nerovnovážné simulace nad a pod teplotou přechodu mezi fluidní a gelovou fází. Díky interakcím alkylových řetízků s hydrofobním povrchem vlasu se vytvořila hustá adsorbovaná vrstva, jejíž struktura se lišila při fluidních a gelových podmínkách. Při aplikaci smykového pohybu, který aproximuje reálné nerovnovážné podmínky, se ve fluidní fázi molekuly alkoholů a surfaktantů desorbovaly úměrně smykové rychlosti, zatímco v gelové fázi desorbce nastala rychle po dosažení prahové smykové rychlosti. Desorbované molekuly pak v roztoku tvořili různé samouspořádané struktury.
Lamely tvořené mastnými alkoholy a nabitými surfaktanty jsou běžnou součástí tekutých kosmetických přípravků. Pomocí mesoskopického modelování jsme studovali, jak monovrstvy pocházející z odpovídajících dvouvrstev v tekutých kosmetických přípravcích interagují s povrchem lidského vlasu. Prováděli jsme rovnovážné i nerovnovážné simulace nad a pod teplotou přechodu mezi fluidní a gelovou fází. Díky interakcím alkylových řetízků s hydrofobním povrchem vlasu se vytvořila hustá adsorbovaná vrstva, jejíž struktura se lišila při fluidních a gelových podmínkách. Při aplikaci smykového pohybu, který aproximuje reálné nerovnovážné podmínky, se ve fluidní fázi molekuly alkoholů a surfaktantů desorbovaly úměrně smykové rychlosti, zatímco v gelové fázi desorbce nastala rychle po dosažení prahové smykové rychlosti. Desorbované molekuly pak v roztoku tvořili různé samouspořádané struktury.
- K. Šindelka, A. Kowalski, M. Cooke, C. Mendoza, M. Lísal, Interactions of cationic surfactant-fatty alcohol monolayers with natural human hair surface: Insights from dissipative particle dynamics, J. Mol. Liq. 375, 121385, 2023. DOI
Tlak vody uvnitř geologických zlomů při nukleaci zemětřesení
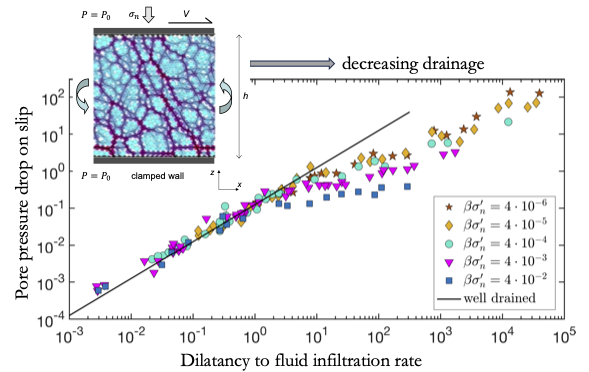 Zemětřesení obvykle nastává skluzem na existujících zlomech v zemské kúře. Takový zlom si lze představit jako tenkou vrstvu granulárního materiálu oddělující dva bloky okolní soudržné horniny. Zlomy jsou často nasáklé vodou, která může jak podpořit, tak utlumit vznik zemětřesení. Například, pokud se zlom, který se zrovna začíná smýkat, roztáhne, tlak vody uvnitř zlomu poklesne, což vyvolá sílu, která tlumí vznikající deformaci (zemětřesení). Měření tohoto efektu vody je však experimentálně prakticky nemožné. Nám se podařilo předpovědět tlak vody uvnitř smýkané granulární vrstvy pomocí fyzikálního modelu a počítačových simulací. Obrázek zobrazuje pokles tlaku vody uvnitř granulární vrstvy, která se začíná smýkat. Tlak je převážně určen počáteční rychlostí smyku a permeabilitou granulární vrstvy, což je parametr, který ovlivňuje rychlost s jakou rozpínající se vrstva nasává vodu z okolní horniny.
Zemětřesení obvykle nastává skluzem na existujících zlomech v zemské kúře. Takový zlom si lze představit jako tenkou vrstvu granulárního materiálu oddělující dva bloky okolní soudržné horniny. Zlomy jsou často nasáklé vodou, která může jak podpořit, tak utlumit vznik zemětřesení. Například, pokud se zlom, který se zrovna začíná smýkat, roztáhne, tlak vody uvnitř zlomu poklesne, což vyvolá sílu, která tlumí vznikající deformaci (zemětřesení). Měření tohoto efektu vody je však experimentálně prakticky nemožné. Nám se podařilo předpovědět tlak vody uvnitř smýkané granulární vrstvy pomocí fyzikálního modelu a počítačových simulací. Obrázek zobrazuje pokles tlaku vody uvnitř granulární vrstvy, která se začíná smýkat. Tlak je převážně určen počáteční rychlostí smyku a permeabilitou granulární vrstvy, což je parametr, který ovlivňuje rychlost s jakou rozpínající se vrstva nasává vodu z okolní horniny.
- S. Parez, M. Kozakovic, J. Havlica, Pore pressure drop during dynamic rupture and conditions for dilatancy hardening, J. Geophys. Res. Solid Earth 128, e2023JB026396, 2023. DOI
Recognition of the granular airborne portion in a flighted rotary drum
This ar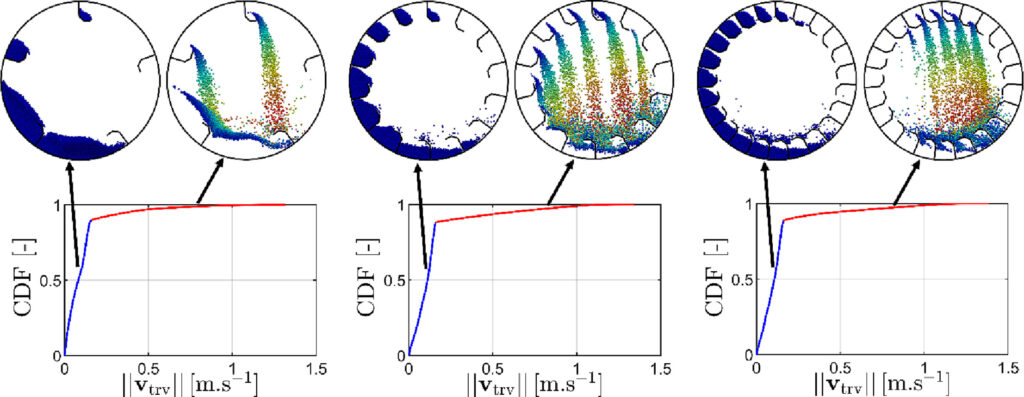 ticle deals with numerical simulation via Discrete Element Method (DEM) in a flighted rotary drum with a varying number of flights from 1 to 25 at a constant load of particles. The granular kinematics is studied in the three regimes: under-, design- and over-loading. It was shown that the behavior of granular material during the unloading process from the flights governs the regime transitions. The cross-sectional distribution analysis of the granular material in the different parts of the drum established the existence of two dense media (bed and flights) and one dilute medium (in the airborne portion). Since one of the criteria that affect the efficiency of heat transport between the gas and the particle media is the number of particles in the airborne phase, three recognition methods are proposed to detect the airborne portion. Based on geometry, velocity magnitude, and minimal separation distance, the comparison between them provides a basis for discussion about the relevance of each method in detecting airborne particles.
ticle deals with numerical simulation via Discrete Element Method (DEM) in a flighted rotary drum with a varying number of flights from 1 to 25 at a constant load of particles. The granular kinematics is studied in the three regimes: under-, design- and over-loading. It was shown that the behavior of granular material during the unloading process from the flights governs the regime transitions. The cross-sectional distribution analysis of the granular material in the different parts of the drum established the existence of two dense media (bed and flights) and one dilute medium (in the airborne portion). Since one of the criteria that affect the efficiency of heat transport between the gas and the particle media is the number of particles in the airborne phase, three recognition methods are proposed to detect the airborne portion. Based on geometry, velocity magnitude, and minimal separation distance, the comparison between them provides a basis for discussion about the relevance of each method in detecting airborne particles.
- M. Kozakovic, J. Havlica, L. Le Guen, S. Parez, F. Huchet, Recognition of the granular airborne portion in a flighted rotary drum, Powder Technology 425, 118565, 2023. DOI
Fázové přechody a tvary kapek nad hydrofilními proužky
Uvažujeme adsorpci tekutiny na rovinné stěně vzorkované proužkem o šířce L hydrofilního materiálu, který je obklopen materiálem ve stavu částečného smáčení. Předpokládáme, že pokud by šířka L byla nekonečná, vykazovala by stěna fázový přechod smáčení prvního řádu při teplotě Tw a odpovídající fázovou křivku předsmáčení. Pro konečné šířky L a) určíme posun konečné velikosti teploty smáčení Tw(L), b) odvodíme rovnici, analogickou Kelvinově rovnici pro kapilární kondenzaci, pro posun křivky předsmáčení, c) ukážeme škálování a konformně invariantní vlastnosti kapek kondenzované kapaliny ležících nad pruhem . Dále poukážeme na analogie mezi těmito předpověďmi a těmi pro kapilární kondenzaci mezi paralelními deskami a tyto výsledky testujeme numericky pomocí mikroskopické nelokální klasické teorie funkcionálu hustoty.
- M. Pospíšil, A. O. Parry, A. Malijevský, Fázové přechody a tvary kapek nad hydrofilními proužky, J. Mol. Liq. 381, 121834, 2023. DOI
Molekulární simulace hydrátů halogenidů alkalických kovů
 Je známo, že klasické molekulární simulace koncentrovaných vodných roztoků elektrolytů jsou omezeny schopností mikroskopických modelů (silových polí) předpovídat správné hodnoty rozpustnosti solí, což vyžaduje i jejich schopnost věrně modelovat krystalické soli. Předchozí simulační studie se často zaměřovaly na rozpustnost bezvodých krystalických solí, ale prakticky nikdy na krystalické hydráty, s výjimkou hydrohalitu, NaCl.2H2O, přestože je experimentálně známo nejméně 23 různých hydrátů, které se mohou srážet z roztoků alkalických halogenidů. Tato práce se pokouší zaplnit tuto mezeru ve studiích simulace hydrátů systematickým zkoumáním schopnosti vybraných nejlepších silových polí kvalitativně zachytit stabilitu jednotlivých fází různých hydrátů alkalických halogenidů a kvantitativně předpovědět jejich mřížkové parametry. Nejprve jsme ukázali, že studovaná nepolarizovatelná silová pole často nedokážou modelovat hydráty obsahující kationty Li+ , zatímco námi nedávno zdokonalená polarizovatelná silová pole jsou schopna modelovat všechny hydráty s výjimkou LiCl.H2O. Za druhé, dále jsme zpřesnili naše silové pole pro Li+ tak, aby poskytovaly stabilní LiCl.H2O. Za třetí naše simulace objasňují polohy kationtů Li+ v fázích LiBr.H2O a LiI.H2O, jejichž rozložení bylo dříve popsáno pouze jako stochastické. Jako vedlejší produkt je navržena a otestována jednoduchá a spolehlivá metodika simulace vhodná i pro složité polarizovatelné modely a neorthombické krystalové mřížky, založená na simulacích konečných krystalů plovoucích ve vakuu.
Je známo, že klasické molekulární simulace koncentrovaných vodných roztoků elektrolytů jsou omezeny schopností mikroskopických modelů (silových polí) předpovídat správné hodnoty rozpustnosti solí, což vyžaduje i jejich schopnost věrně modelovat krystalické soli. Předchozí simulační studie se často zaměřovaly na rozpustnost bezvodých krystalických solí, ale prakticky nikdy na krystalické hydráty, s výjimkou hydrohalitu, NaCl.2H2O, přestože je experimentálně známo nejméně 23 různých hydrátů, které se mohou srážet z roztoků alkalických halogenidů. Tato práce se pokouší zaplnit tuto mezeru ve studiích simulace hydrátů systematickým zkoumáním schopnosti vybraných nejlepších silových polí kvalitativně zachytit stabilitu jednotlivých fází různých hydrátů alkalických halogenidů a kvantitativně předpovědět jejich mřížkové parametry. Nejprve jsme ukázali, že studovaná nepolarizovatelná silová pole často nedokážou modelovat hydráty obsahující kationty Li+ , zatímco námi nedávno zdokonalená polarizovatelná silová pole jsou schopna modelovat všechny hydráty s výjimkou LiCl.H2O. Za druhé, dále jsme zpřesnili naše silové pole pro Li+ tak, aby poskytovaly stabilní LiCl.H2O. Za třetí naše simulace objasňují polohy kationtů Li+ v fázích LiBr.H2O a LiI.H2O, jejichž rozložení bylo dříve popsáno pouze jako stochastické. Jako vedlejší produkt je navržena a otestována jednoduchá a spolehlivá metodika simulace vhodná i pro složité polarizovatelné modely a neorthombické krystalové mřížky, založená na simulacích konečných krystalů plovoucích ve vakuu.
- P. Matysová, M. Lísal, F. Moučka, Molecular simulations of alkali metal halide hydrates, J. Mol. Liq. 384, 122197, 2023. DOI
Transportní koeficienty z Einsteinových-Helfandových vztahů pomocí standardní a isoenergetické disipativní částicové dynamiky
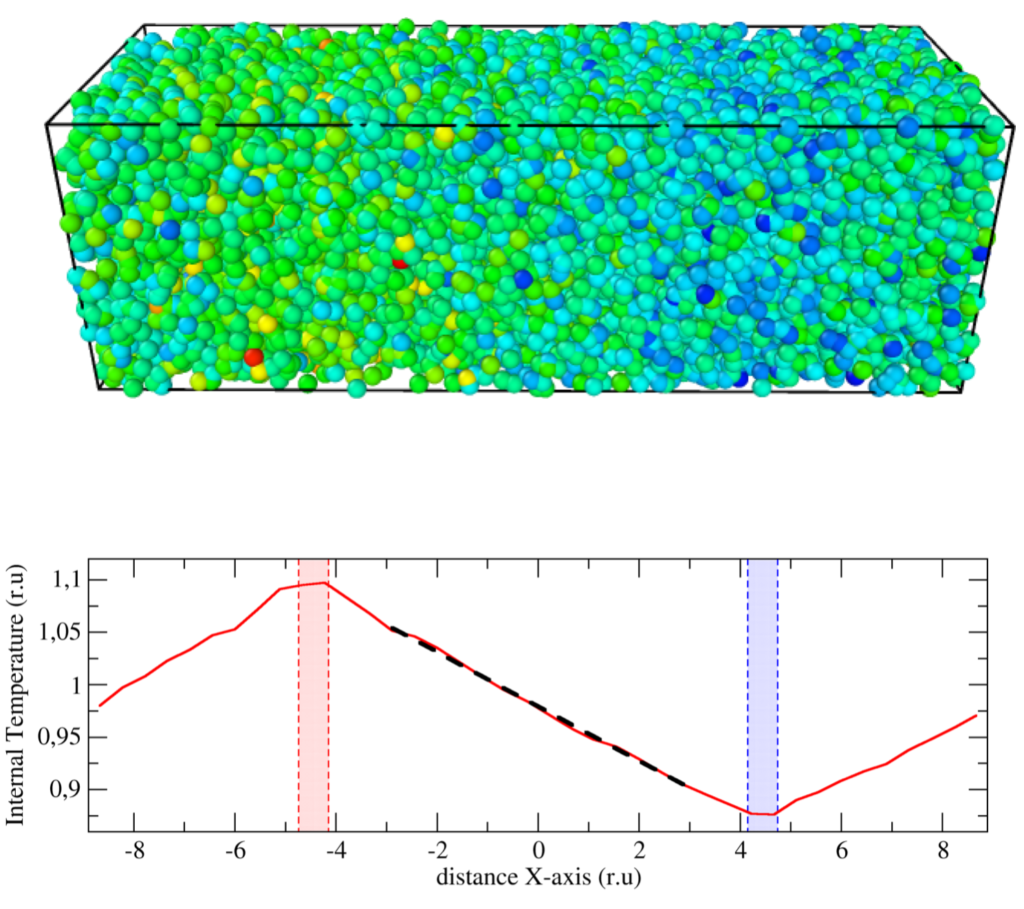 Ukázali jsme, že navzdory obecnému přesvědčení jsou standardní Einsteinovy-Helfandovy (EH) vzorce platné pro vyhodnocení transportních koeficientů systémů obsahujících disipativní a náhodné síly za předpokladu, že pro tyto mezoskopické systémy jsou: (i) splněny příslušné zákony zachování a (ii) pravděpodobnosti přechodu splňují princip detailní rovnováhy. Příkladem takových mesoskopických metod jsou například disipativní dynamika částic (DPD) a isoenergetické metody DPD (DPDE). Pro ověření tohoto tvrzení jsme odvodili mezoskopický tvar tepelného toku pro metodu DPDE, vhodný pro výpočet tepelné vodivosti pomocí EH vztahů. Porovnali jsme EH metodu s nerovnovážnými simulacemi pro různé scénáře, včetně vícečásticových potenciálů, a ve všech případech jsme zjistili vynikající shodu. EH vztahy jsou platné zejména pro systémy s potenciály závislými na hustotě a teplotě, jako je nedávno vyvinutá zobecněná metoda DPDE (GenDPDE) [Avalos et al., Phys. Chem. Chem. Phys. 21, 24891, 2019].
Ukázali jsme, že navzdory obecnému přesvědčení jsou standardní Einsteinovy-Helfandovy (EH) vzorce platné pro vyhodnocení transportních koeficientů systémů obsahujících disipativní a náhodné síly za předpokladu, že pro tyto mezoskopické systémy jsou: (i) splněny příslušné zákony zachování a (ii) pravděpodobnosti přechodu splňují princip detailní rovnováhy. Příkladem takových mesoskopických metod jsou například disipativní dynamika částic (DPD) a isoenergetické metody DPD (DPDE). Pro ověření tohoto tvrzení jsme odvodili mezoskopický tvar tepelného toku pro metodu DPDE, vhodný pro výpočet tepelné vodivosti pomocí EH vztahů. Porovnali jsme EH metodu s nerovnovážnými simulacemi pro různé scénáře, včetně vícečásticových potenciálů, a ve všech případech jsme zjistili vynikající shodu. EH vztahy jsou platné zejména pro systémy s potenciály závislými na hustotě a teplotě, jako je nedávno vyvinutá zobecněná metoda DPDE (GenDPDE) [Avalos et al., Phys. Chem. Chem. Phys. 21, 24891, 2019].
- C. Malaspina, M. Lísal, J. P. Larentzos, J. K. Brennan, A. D. Mackie, J. Bonet Avalos, Transport coefficients from Einstein-Helfand relations using standard and energy-conserving dissipative particle dynamics methods, Phys. Chem. Chem. Phys. 25, 12025-12040, 2023. DOI
Adsorpce, difuze a transport alkanů C1 až C3 a oxidu uhličitého v kerogenech s duální porozitou: Poznatky z molekulárních simulací
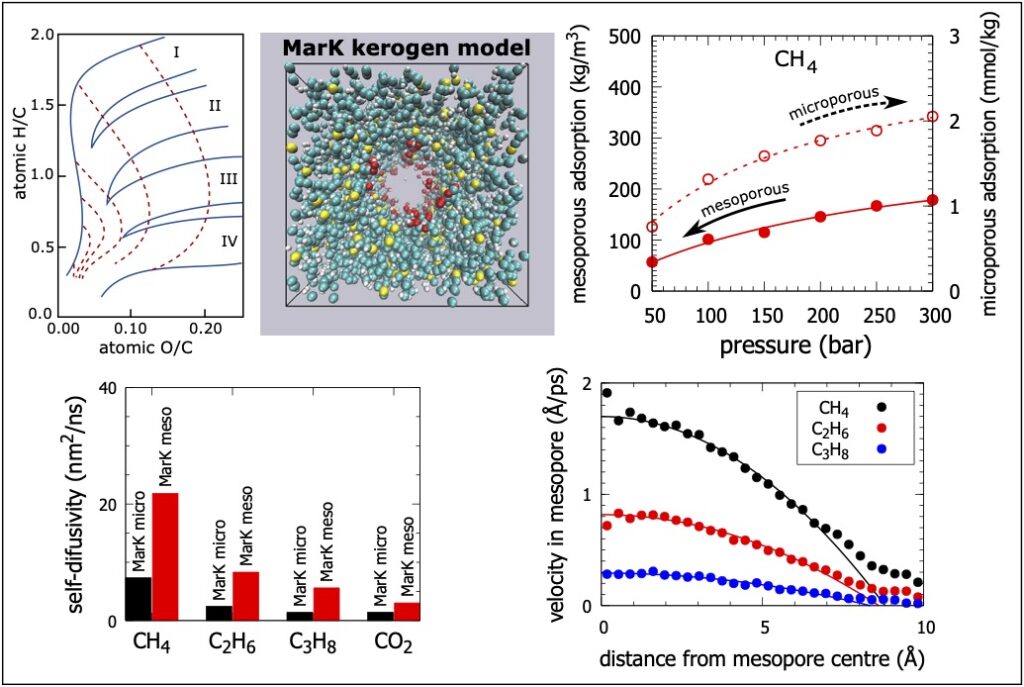 Organické břidlice jsou nekonvenční zásobníky plynu s širokým rozdělením velikosti pórů. Břidlice se skládají ze dvou odlišných složek: organické hmoty a jílových minerálů. Velikost pórů v organické hmotě je většinou soustředěna na méně než 6 nanometrů a tyto mikropóry a malé mezopóry poskytují většinu adsorpční plochy a objemu zásob plynu. V těchto pórech o velikosti nanometrů se chování geo-tekutin výrazně liší od chování v objemové fázi v důsledku silných interakcí mezi pevnou látkou a tekutinou jako i dalších efektů v nanopórech. Pochopení vlastností tekutin jako je metan, etan, propan a oxid uhličitý v úzkých břidlicových pórech má zásadní význam na hydraulické frakování břidlicového plynu a minimalizaci jeho negativních vlivů na životní prostředí. Dále se pro zvýšenou těžbu břidlicového plynu používá oxid uhličitý. Na molekulární úrovni jsme simulovali adsorpci, difuzi a transport metanu, ethanu, propanu a oxidu uhličitého v realistických modelech organických břidlicových materiálů při typických teplotách a tlacích v břidlicovém ložisku. Nejprve jsme použili hybridní reverzní Monte Carlo spolu s experimentálními párovými distribučními funkcemi k vytvoření duálně-porézních kerogenních modelů odpovídajících kerogenům z ložiska Eagle Ford a Marcellus bohatého na jíly. Poté jsme použili grandkanonické Monte Carlo simulace ke studiu adsorpce tekutin v porézních kerogenních strukturách. Adsorpční studie jsme doplňili simulací auto-, kolektivní a transportní difuze molekul adsorbovaných tekutin v břidlicových kerogenech pomocí rovnovážné a nerovnovážné molekulární dynamiky.
Organické břidlice jsou nekonvenční zásobníky plynu s širokým rozdělením velikosti pórů. Břidlice se skládají ze dvou odlišných složek: organické hmoty a jílových minerálů. Velikost pórů v organické hmotě je většinou soustředěna na méně než 6 nanometrů a tyto mikropóry a malé mezopóry poskytují většinu adsorpční plochy a objemu zásob plynu. V těchto pórech o velikosti nanometrů se chování geo-tekutin výrazně liší od chování v objemové fázi v důsledku silných interakcí mezi pevnou látkou a tekutinou jako i dalších efektů v nanopórech. Pochopení vlastností tekutin jako je metan, etan, propan a oxid uhličitý v úzkých břidlicových pórech má zásadní význam na hydraulické frakování břidlicového plynu a minimalizaci jeho negativních vlivů na životní prostředí. Dále se pro zvýšenou těžbu břidlicového plynu používá oxid uhličitý. Na molekulární úrovni jsme simulovali adsorpci, difuzi a transport metanu, ethanu, propanu a oxidu uhličitého v realistických modelech organických břidlicových materiálů při typických teplotách a tlacích v břidlicovém ložisku. Nejprve jsme použili hybridní reverzní Monte Carlo spolu s experimentálními párovými distribučními funkcemi k vytvoření duálně-porézních kerogenních modelů odpovídajících kerogenům z ložiska Eagle Ford a Marcellus bohatého na jíly. Poté jsme použili grandkanonické Monte Carlo simulace ke studiu adsorpce tekutin v porézních kerogenních strukturách. Adsorpční studie jsme doplňili simulací auto-, kolektivní a transportní difuze molekul adsorbovaných tekutin v břidlicových kerogenech pomocí rovnovážné a nerovnovážné molekulární dynamiky.
- E. Rezlerová, S. K. Jain, M. Lísal, Adsorption, diffusion, and transport of C1 to C3 alkanes and carbon dioxide in dual-porosity kerogens: Insights from molecular simulations, Energy Fuels 37, 492-508, 2023. DOI